2011年の総括
私は今まで、この「ショートリードの憂鬱」というブログで、次世代シーケンサーの技術と解析を中心に書いてきました。
仕事でNGSのデータを扱っていましたが、このブログは会社の業務とは全く別に、個人でやっています。
だから自分の所属する会社名は出していないはず(リンクを貼ったことはあったけど)です。
そもそも次世代シーケンサーなるものを最初に知ったのは、2007年の分子生物学会でした。
確かABI社のSOLiDのプレゼンだったと記憶しています。
「これは来るぞ!」って直感し、翌2008年から、メーカーさん主催のセミナーやWebセミナーにはできる限り参加して、情報収集をしていました。
でもまだその頃は、データ量の多さと、将来の可能性ばかりが強調されて、解析はこれから、という感じでした。少なくとも日本では。
2009年、NGS関連の論文がたくさん出てくるようになり、時間が許す限り、目を通すようにしました。
シーケンサーといっても、配列解析だけでなく、発現やメチレーションなど、汎用性が高いのが特徴なので、はっきり言って私の知らない分野が多く、勉強するのが大変です。
この頃からフリーツールを調査し始めるようになります(まだ趣味の範囲で)。
2010年、仕事でNGSのデータを扱うようになって、いよいよ本格的に、世に出ているフリーツールを試し始めました。
また同時に、会社で導入したCLC-Bio社のソフトを触るうちに、他の商用ソフトとの比較もしたくなり、いくつかトライアルしました。
そんな経験やそこで得た情報を何とか残しておきたい、ほかのひととシェアしたい、と始めたのがこのブログです。
開設から1年半経ち、このブログがきっかけで、出会えたひともたくさんいます。
本当に良かったと思います。
本日12月29日は、会社の仕事納め、と同時に私の最後の出勤日でした。
来年1月からは、Pacific Biosciences社の日本代理店で働きます!
PacBioについては前から何回か書いてきましたね。 噂の一分子のシーケンサーです。
そこで、
PacBioのことは別ブログで書くことにしました。 もうショートリードじゃないし・・・
その名も、
「パックマンの挑戦」
PacBioにとっても、私にとっても、2012年は挑戦です。
日本で一番詳しいPacBioのブログ、を目指します!
とにかく、PacBioのシーケンサーは、賛否両論、いろいろ言われていますね。
それだけ注目度が高いということでしょう。
新しい技術は、批判に聞く耳を持って、改善されて、世に広がっていくものです。
私は、今度はPacBio擁護派に属する訳ですが、そのブログでは、正確な情報を発信することに気をつけます。
つまり、批判が正しければそのように、間違っていればその点を明らかにしようと思います。
また、PacBioに興味がある方全てに向けて、公共のPacBioデータとネット上の情報の、解説っぽいことも書こうと思います。
それから、PacBioのデータ解析も、いち早く書こうと思います。
と、まあ、来年に向けての抱負っぽいことを言いましたが、どれだけ実現できるか・・・
「ショートリードの憂鬱」も、できる限り、続けていくつもりです。
主に公共にある情報(論文やネット上)が中心になるかと思いますが。
やはりロングリード(特に一分子)をやる以上は、ライバルのショートリードを知ることが欠かせないですから。
来年もどうぞ宜しくお願いします。
良いお年を!!
2011年12月27日火曜日
BEDファイルをGFFファイルに変換
BEDフォーマットからGFFフォーマットへの変換
って、何のために?
と思った方もいるのではないでしょうか。
どちらも、ゲノムのアノテーションや場所を指定・定義するフォーマットです。
例えば前回のブログに書いた、SureSelectのキャプチャー領域を示したBEDファイル、これをGFFファイルに変換する必要があったとします。
(GFFしか対応していないツールにBEDファイルの情報を取り込みたい時など)
フォーマットを単に変換すれば良いと思いきや、注意する点がありました。
ポジションの表示方法が、UCSCで作られたBEDフォーマットは0インデックス、SangerでつくられたGFFは1インデックスなのです。
なんのこっちゃ?とお思いの方、数え方の違いだと思ってください。
地上階を1階と数えるか、(行ったことありませんがイギリスのように)Ground Floorとして0階と数えるか、の違いのような。
塩基を0から数えるか、1から数えるか、の差です。
くわしくはここを参照。
プログラムでやるのも良いですが、車輪の再発明、なんてことにならないように、もっと簡単便利なツールがGalaxyです。
Galaxyはこちら ユーザー登録が必要です。
先ずはデータ(BEDファイル)をアップロードします。

File Formatはbedを選択、ファイル(ここではSureSelect のHuman Exon 50MbのBedファイル)を指定、GenomeはHg19で、Executeボタンを押します。
ネットワークによっては結構時間がかかりますので、気長に待ちます。
うまくアップロードされると右側のパネルに、ファイル名が表示されます。

Galaxyはいろいろできるクラウド型解析パイプラインです。
個人的には、フォーマットのコンバート(変換)など、ちょっとしたことに使っています。
その中のひとつ、BED-to-GFFを使います。
左側パネル、Convert Formats をクリックし、Convert this query に今アップしたSureSelectのBedファイルを選択して Execute します。

これはすぐ終わるでしょう。 GFFファイルができたら、保存アイコンを押してダウンロード。

では、早速、GFFファイルを確認してみましょう。
中身を見るのももちろんですが、ソフトにインポートしてうまく入るかどうかを確認して下さい。
IGVに入れたときの例:
下段の一番上がRefSeqのアノテーション、真ん中がGFF変換後のSureSelect Human 50Mb、下が変換前のBEDファイル。
真ん中と一番下は同じ情報のはずです。
 もっと拡大して塩基がずれていないか確認。
もっと拡大して塩基がずれていないか確認。
 ずれていないようですね。
ずれていないようですね。
ちなみに、BEDファイルとGFFファイルは、IGVに取り込む前に、File>Run igvtoolsでIGVtoolsを開き、Indexを付けてあげる作業が必要かもしれません。
さて、最初に戻り、なんでこんな、わざわざBEDで書いてあるアノテーションをGFFにしたか、という理由をお話します。
あるソフト、CLC Genomics Workbenchでは、リファレンス配列にアノテーションを追加する機能がありますが、現バージョン4.9ではBEDファイルを取り込めないのです。
GFFならOK、ということで、わざわざ同じ情報をGFFにしたのでした。
こうすると、リファレンスゲノムにSureSelectのキャプチャー領域がアノテーションされるので、Exome実験のデータをマッピングするときに、SureSelectキャプチャー領域のみに、マップすることが可能になるのです。
もちろんSureSelectだけでなく、TruSeqやSeqCapなどでキャプチャーしても同じです。 BEDファイルがあればGFFに変換してCLCに取り込めば、キャプチャー領域だけにマッピングすることができます。
それがやりたかったのでした。
って、何のために?
と思った方もいるのではないでしょうか。
どちらも、ゲノムのアノテーションや場所を指定・定義するフォーマットです。
例えば前回のブログに書いた、SureSelectのキャプチャー領域を示したBEDファイル、これをGFFファイルに変換する必要があったとします。
(GFFしか対応していないツールにBEDファイルの情報を取り込みたい時など)
フォーマットを単に変換すれば良いと思いきや、注意する点がありました。
ポジションの表示方法が、UCSCで作られたBEDフォーマットは0インデックス、SangerでつくられたGFFは1インデックスなのです。
なんのこっちゃ?とお思いの方、数え方の違いだと思ってください。
地上階を1階と数えるか、(行ったことありませんがイギリスのように)Ground Floorとして0階と数えるか、の違いのような。
塩基を0から数えるか、1から数えるか、の差です。
くわしくはここを参照。
プログラムでやるのも良いですが、車輪の再発明、なんてことにならないように、もっと簡単便利なツールがGalaxyです。
Galaxyはこちら ユーザー登録が必要です。
先ずはデータ(BEDファイル)をアップロードします。

File Formatはbedを選択、ファイル(ここではSureSelect のHuman Exon 50MbのBedファイル)を指定、GenomeはHg19で、Executeボタンを押します。
ネットワークによっては結構時間がかかりますので、気長に待ちます。
うまくアップロードされると右側のパネルに、ファイル名が表示されます。

Galaxyはいろいろできるクラウド型解析パイプラインです。
個人的には、フォーマットのコンバート(変換)など、ちょっとしたことに使っています。
その中のひとつ、BED-to-GFFを使います。
左側パネル、Convert Formats をクリックし、Convert this query に今アップしたSureSelectのBedファイルを選択して Execute します。

これはすぐ終わるでしょう。 GFFファイルができたら、保存アイコンを押してダウンロード。

では、早速、GFFファイルを確認してみましょう。
中身を見るのももちろんですが、ソフトにインポートしてうまく入るかどうかを確認して下さい。
IGVに入れたときの例:
下段の一番上がRefSeqのアノテーション、真ん中がGFF変換後のSureSelect Human 50Mb、下が変換前のBEDファイル。
真ん中と一番下は同じ情報のはずです。


ちなみに、BEDファイルとGFFファイルは、IGVに取り込む前に、File>Run igvtoolsでIGVtoolsを開き、Indexを付けてあげる作業が必要かもしれません。
さて、最初に戻り、なんでこんな、わざわざBEDで書いてあるアノテーションをGFFにしたか、という理由をお話します。
あるソフト、CLC Genomics Workbenchでは、リファレンス配列にアノテーションを追加する機能がありますが、現バージョン4.9ではBEDファイルを取り込めないのです。
GFFならOK、ということで、わざわざ同じ情報をGFFにしたのでした。
こうすると、リファレンスゲノムにSureSelectのキャプチャー領域がアノテーションされるので、Exome実験のデータをマッピングするときに、SureSelectキャプチャー領域のみに、マップすることが可能になるのです。
もちろんSureSelectだけでなく、TruSeqやSeqCapなどでキャプチャーしても同じです。 BEDファイルがあればGFFに変換してCLCに取り込めば、キャプチャー領域だけにマッピングすることができます。
それがやりたかったのでした。
2011年12月26日月曜日
SureSelect のBEDファイル取得方法
Exome解析については、以前ここでも書きましたが、ワークフローは大体出来上がっています。
NGS Surfer's Wiki のリシークエンス
BioStar を参照
このうち、3の、キャプチャーした領域だけを取り出す、という所は、キャプチャー領域を定義したファイルが必要です。
このファイル、BEDフォーマットであることが普通(Bedtoolsを使うときは)です。
UCSCなどの公共DBから落とすことも可能でしょうが、メーカーももちろん提供しています。
Agilent社のSureSelect Human Whole Exon 50MのBEDファイルをダウンロードして、実際にどの場所をキャプチャーしているのか、見てみましょう。
Agilent社のアレイ情報は、eArray というサイトで得ることができます。 登録が必要ですが、無料なので是非ユーザー登録しましょう!
ログイン画面はこんな感じ
 ちょっと重いサイトですが、気にせずに。
ちょっと重いサイトですが、気にせずに。
 右上の、Switch Application Type をクリックして、
右上の、Switch Application Type をクリックして、
 SureSelect Target Enrichment を選びます。
SureSelect Target Enrichment を選びます。
 Human All Exon 50Mb Kit というのが欲しいアレイ情報です。
Human All Exon 50Mb Kit というのが欲しいアレイ情報です。
これをダウンロードしましょう!
ゲノムバージョンを確認して、ダウンロード画面へ
 BEDだけをダウンロードするのではなく、ここでは全部選択します。
BEDだけをダウンロードするのではなく、ここでは全部選択します。
欲しいのは、実は一番上のSureSelect_All_Exon・・・というファイルなのです。
後で中身を比較してみると良いでしょう。

ダウンロードが終わったら、ファイルを解凍して、さらにSureSelect_All_Exon・・・.zipを解凍して、.bedファイルを得ます。 このBedファイルはキャプチャー領域を定義しています。
先のBEDフォルダに入っているBedファイルは、プローブの設定領域を定義しているようです。
Bedファイルの中を開くとこんな感じ

これをIGV (Integrative Genomics Viewer http://www.broadinstitute.org/igv/ )上で見てみましょう!
先ず、IGVtools でインデックスを付ける必要があります。
(File > Run igvtools... でBedファイルを指定し、Indexを選択)
これをインポートすればOK!
画像は次回、このBedファイルと、BedファイルをGalaxyというフリーツールを使ってGFFファイルに変換する方法を説明するときに合わせてお見せします。
では。
- リードをゲノムにマッピングし (BWA)
- 冗長性のあるリード・Duplicateを除去し (SamtoolsやPicard)
- キャプチャーしたExon領域だけを取り出し (Bedtools)
- SamtoolsでSNVを抽出し
- SNVにアノテーションをつける non-synonymous SNV, missense, frame-shift など
NGS Surfer's Wiki のリシークエンス
BioStar を参照
このうち、3の、キャプチャーした領域だけを取り出す、という所は、キャプチャー領域を定義したファイルが必要です。
このファイル、BEDフォーマットであることが普通(Bedtoolsを使うときは)です。
UCSCなどの公共DBから落とすことも可能でしょうが、メーカーももちろん提供しています。
Agilent社のSureSelect Human Whole Exon 50MのBEDファイルをダウンロードして、実際にどの場所をキャプチャーしているのか、見てみましょう。
Agilent社のアレイ情報は、eArray というサイトで得ることができます。 登録が必要ですが、無料なので是非ユーザー登録しましょう!
ログイン画面はこんな感じ




これをダウンロードしましょう!
ゲノムバージョンを確認して、ダウンロード画面へ

欲しいのは、実は一番上のSureSelect_All_Exon・・・というファイルなのです。
後で中身を比較してみると良いでしょう。

ダウンロードが終わったら、ファイルを解凍して、さらにSureSelect_All_Exon・・・.zipを解凍して、.bedファイルを得ます。 このBedファイルはキャプチャー領域を定義しています。
先のBEDフォルダに入っているBedファイルは、プローブの設定領域を定義しているようです。
Bedファイルの中を開くとこんな感じ

これをIGV (Integrative Genomics Viewer http://www.broadinstitute.org/igv/ )上で見てみましょう!
先ず、IGVtools でインデックスを付ける必要があります。
(File > Run igvtools... でBedファイルを指定し、Indexを選択)
これをインポートすればOK!
画像は次回、このBedファイルと、BedファイルをGalaxyというフリーツールを使ってGFFファイルに変換する方法を説明するときに合わせてお見せします。
では。
2011年12月20日火曜日
キメラRNAの検出
NGSでキメラRNAの検出に挑戦した論文
Kannal et al. Recurrent chimeric RNAs enriched in human prostate cancer identified by deep sequencing PubMed

Q: Chimeric RNAって、Fusion Geneのことでしょうか?
A: もっと広い概念です。 Chimeric RNAのうち、Chromosomal Rearrangementsによって作られる複合遺伝子のことを、Fusion Geneと定義するそうです。
染色体リアレンジメント以外の原因で、転写が原因で作られるキメリックな転写産物のことも、Chimeric RNAと呼ぶそうです。
転写が原因で起こる?
そのうち、前立腺癌に特徴的なキメラRNAを見つけよう!という論文です。
NGSはスクリーニングに使う、というスタンスです。
いくつもフィルタリングを重ねて、キメラRNAの候補を絞り込んでいます。


Kannal et al. Recurrent chimeric RNAs enriched in human prostate cancer identified by deep sequencing PubMed

Q: Chimeric RNAって、Fusion Geneのことでしょうか?
A: もっと広い概念です。 Chimeric RNAのうち、Chromosomal Rearrangementsによって作られる複合遺伝子のことを、Fusion Geneと定義するそうです。
染色体リアレンジメント以外の原因で、転写が原因で作られるキメリックな転写産物のことも、Chimeric RNAと呼ぶそうです。
転写が原因で起こる?
- 隣の下流遺伝子まで、転写されてしまうことで作られる、Read-through/splicing 型Chimeric RNA
- 転写された異なるRNA分子が同時にスプライシングされて、ひとつのRNA分子になる、Transsplicing 型Chimeric RNA
そのうち、前立腺癌に特徴的なキメラRNAを見つけよう!という論文です。
NGSはスクリーニングに使う、というスタンスです。
いくつもフィルタリングを重ねて、キメラRNAの候補を絞り込んでいます。
イルミナGAIIの36bpペアエンドリードを使っています。

- クオリティフィルター: リード単位でクオリティが悪いもの(イルミナパイプラインのデフォルト)を除きます
- ユニークネスフィルター: リードマッピングの後、マッパブルなペアエンドリードのうち、ペアとして、ゲノムまたはTranscriptに複数個所マップするものを除きます。 これによって、ペアリードの両方とも、ゲノム上またはTranscript上に1か所だけマッピングするものだけが残ります(2塩基のミスマッチはゆるしています)。
- Entrez Gene IDフィルター: ゲノムやTranscriptにユニークにヒットすると言っても、遺伝子単位でユニークかどうか。 彼らは、Entrez Gene ID単位で遺伝子を定義しています(Gene IDが無い配列は遺伝子としない)。 ペアとして複数のGene IDにマップされるリードは除かれます。 この2と3のフィルタリングは、ユニークマッピングするペアリードだけを残すので、Gene Family(Homolog)などを間違ってキメラRNA候補としてしまうのを避けることができます。 しかし、逆に、Homologのどれかがキメラっている場合のRNAは検出できません。 実際、CLC Genomics Workbench でも経験があるのですが、Fusion Geneを検出してもHomologにばかりヒットして、擬陽性ばかりということがありました。 ユニークヒットに限ることは良いかもしれません。
- Gene ID キメラフィルター: キメラRNAを見つけるのが目的なので、同じ遺伝子上にペアの両方ともマップされているリードは除きます。 異なる遺伝子をまたいでマップされているリードだけに絞っています。 イメージでは、こんな感じ(Fusion Geneと書いていますがキメラRNAと置き換えてください)

ペアの片方が、偶然GeneAとGeneBの結合部位(ジャンクション)に合った場合、片方のペアの6塩基が完全ヒットであれば、ジャンクションにヒットしたと、見なしていました。
個人的にはもう少し長い塩基の条件にした方が良いのでは、と。
彼らは、少なくとも3本のペアリードが、同じキメラRNAの相手にマップされていた、という条件で、 6,163ものキメラ候補を3人の患者サンプルから得ました。
そのうち、後に検証しにくいKLK2またはKLK3を含むキメラを除くと、2,369にまで減りました。
20人の患者中10人に共通していたキメラは46と、ウェットで検証可能な数になりました。
世の中には、TopHat Fusionというツールがありますが、この論文では使われていないようです。
個人的にはもう少し長い塩基の条件にした方が良いのでは、と。
彼らは、少なくとも3本のペアリードが、同じキメラRNAの相手にマップされていた、という条件で、 6,163ものキメラ候補を3人の患者サンプルから得ました。
そのうち、後に検証しにくいKLK2またはKLK3を含むキメラを除くと、2,369にまで減りました。
20人の患者中10人に共通していたキメラは46と、ウェットで検証可能な数になりました。
世の中には、TopHat Fusionというツールがありますが、この論文では使われていないようです。
2011年12月15日木曜日
分子生物学会 in 横浜
例年通り、分子生物学会に来ています。
展示会のブースに立っていて、色んな人と話をしました。
やっぱり次世代関係の質問は多いです。
昨年に比べて3、4倍くらい、質問がきます。
やってみたい、というひとを入れると10倍くらいに増えているような感じです。
私のブログを見てくれているひともいました。
「もしかしてブログの・・・」 って言ってくれたひとも。 うれしい。
ポスターも、NGS関連が多くなってきたと実感します。
来年の、「NGS現場の会 第二回研究会」のチラシです。
 行かれる方も多いのではないでしょうか?
行かれる方も多いのではないでしょうか?
この分野はまだ始まったばかりですので、手探り状態で実験・解析をしている方が多いのではないかと思います。
「現場の会」のようなところで、意見交換、質問、発表をすることは良いでしょうね。
NGS専門の学会が、日本にはありませんから。
私も来年、行きます。
それにしても横浜は疲れる。
私の家は東京の板橋区なんですが、三田線、東横線、みなとみらい線と乗り継いで、会場のパシフィコ横浜までドア・トゥー・ドアで約2時間。
往復4時間はしんどいです。
神戸が会場の去年は、泊まりだったから、ホテルに帰ってもまだまだ元気だったのに・・・。
展示会のブースに立っていて、色んな人と話をしました。
やっぱり次世代関係の質問は多いです。
昨年に比べて3、4倍くらい、質問がきます。
やってみたい、というひとを入れると10倍くらいに増えているような感じです。
私のブログを見てくれているひともいました。
「もしかしてブログの・・・」 って言ってくれたひとも。 うれしい。
ポスターも、NGS関連が多くなってきたと実感します。
来年の、「NGS現場の会 第二回研究会」のチラシです。

この分野はまだ始まったばかりですので、手探り状態で実験・解析をしている方が多いのではないかと思います。
「現場の会」のようなところで、意見交換、質問、発表をすることは良いでしょうね。
NGS専門の学会が、日本にはありませんから。
私も来年、行きます。
それにしても横浜は疲れる。
私の家は東京の板橋区なんですが、三田線、東横線、みなとみらい線と乗り継いで、会場のパシフィコ横浜までドア・トゥー・ドアで約2時間。
往復4時間はしんどいです。
神戸が会場の去年は、泊まりだったから、ホテルに帰ってもまだまだ元気だったのに・・・。
2011年11月28日月曜日
GRCh37とHg19
突然ですがヒトのゲノムにマッピングをするとき、NCBIの配列を使いますか? それともUCSCの配列を使いますか?
GRCh37 (Genome Reference Consortium human build 37) ?
Hg19 (UCSC human genome 19) ?
リファレンス配列、1番から22番+X、Y染色体に関して言えば、配列はどちらも同じです。
そう言われても信じない方もいると思うし、現にヒューマンエラーで多少違うんでは?と思っている方もいるでしょう。
どちらも全く同じ配列だ、ということはUCSCのゲノムブラウザなどで、GRCh37/hg19 と記述されていることから何となく信じている方がほとんどだと思います。
でも、たまにお客さんから、どっちがいいの? 本当に同じ配列なの? と聞かれることもありました。
そういう疑問を持つ方もいるらしく、私はとりあえず、A、T、C、GそれとNの塩基の数がGRCh37とhg19で同じかどうか、染色体ごとに調べてみました。
「とりあえず」というのは、本当は塩基の並び順も調べるべきなのでしょうが、4つの塩基+Nの数が全く同じなら「合っている」としてしまおう、という大雑把な試みだからです。
GRCh37の配列は
ftp://ftp.ncbi.nih.gov/genomes/H_sapiens/Assembled_chromosomes/seq/
から、hs_ref_GRCh37.p5_chr1.fa.gzなどを1番から22番+XとY
Hg19の配列は
http://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/
から、chr1.fa.gz などを1番から22番+XとY
解凍してfastaファイルにしたら、以下のようなコマンドを実行して塩基の種類と数を得ました。
参考にしたのはこのサイトです。
# UCSCのchr1の場合
tail -n+2 chr1.fa | awk '{ for ( i=1; i<=length; i++ ) arr[substr($0, i, 1)]++ }END{ for ( i in arr ) { print i, arr[i] } }' > hg19_01.txt
A 32485284
N 23970000
C 23064132
T 32559153
G 23070958
a 33085607
c 23960280
g 23945604
t 33109603
# GRCh37のhs_ref_GRCh37.p2_chr1の場合(GRCh37.p5ではなくて.p2でしたね、私が落とした時は。でも同じです。)
tail -n+2 hs_ref_GRCh37.p2_chr1.fa | awk '{ for ( i=1; i<=length; i++ ) arr[substr($0, i, 1)]++ }END{ for ( i in arr ) { print i, arr[i] } }' > GRCh37_01.txt
A 65570891
N 23970000
C 47024412
G 47016562
T 65668756
UCSCのは、塩基の大文字と小文字に分かれていますが、足すとGRCの数と一致することがわかります。
私はこれを、1番から22番+X、Yまで順番に実行して足し算して比べました。
(暇人ではないですよ)
結果、A、T、C、G、Nの数は、3番染色体を除く全ての染色体でUCSCのとGRCのとが一致しました。
唯一つ、3番染色体は、Nの数が違ったのです。
正確には、
60,830,534番目の塩基が、UCSCではN、GRCではM
60,830,763番目と次の塩基が、UCSCではNN、GRCではRR
でした。
以下、ViewerはCLCのGenomics Workbenchを使用

上段がUCSC hg19、下段がGRCh37の3番染色体
NとMが違います。




GRCh37 (Genome Reference Consortium human build 37) ?
Hg19 (UCSC human genome 19) ?
リファレンス配列、1番から22番+X、Y染色体に関して言えば、配列はどちらも同じです。
そう言われても信じない方もいると思うし、現にヒューマンエラーで多少違うんでは?と思っている方もいるでしょう。
どちらも全く同じ配列だ、ということはUCSCのゲノムブラウザなどで、GRCh37/hg19 と記述されていることから何となく信じている方がほとんどだと思います。
でも、たまにお客さんから、どっちがいいの? 本当に同じ配列なの? と聞かれることもありました。
そういう疑問を持つ方もいるらしく、私はとりあえず、A、T、C、GそれとNの塩基の数がGRCh37とhg19で同じかどうか、染色体ごとに調べてみました。
「とりあえず」というのは、本当は塩基の並び順も調べるべきなのでしょうが、4つの塩基+Nの数が全く同じなら「合っている」としてしまおう、という大雑把な試みだからです。
GRCh37の配列は
ftp://ftp.ncbi.nih.gov/genomes/H_sapiens/Assembled_chromosomes/seq/
から、hs_ref_GRCh37.p5_chr1.fa.gzなどを1番から22番+XとY
Hg19の配列は
http://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/
から、chr1.fa.gz などを1番から22番+XとY
解凍してfastaファイルにしたら、以下のようなコマンドを実行して塩基の種類と数を得ました。
参考にしたのはこのサイトです。
# UCSCのchr1の場合
tail -n+2 chr1.fa | awk '{ for ( i=1; i<=length; i++ ) arr[substr($0, i, 1)]++ }END{ for ( i in arr ) { print i, arr[i] } }' > hg19_01.txt
A 32485284
N 23970000
C 23064132
T 32559153
G 23070958
a 33085607
c 23960280
g 23945604
t 33109603
# GRCh37のhs_ref_GRCh37.p2_chr1の場合(GRCh37.p5ではなくて.p2でしたね、私が落とした時は。でも同じです。)
tail -n+2 hs_ref_GRCh37.p2_chr1.fa | awk '{ for ( i=1; i<=length; i++ ) arr[substr($0, i, 1)]++ }END{ for ( i in arr ) { print i, arr[i] } }' > GRCh37_01.txt
A 65570891
N 23970000
C 47024412
G 47016562
T 65668756
UCSCのは、塩基の大文字と小文字に分かれていますが、足すとGRCの数と一致することがわかります。
私はこれを、1番から22番+X、Yまで順番に実行して足し算して比べました。
(暇人ではないですよ)
結果、A、T、C、G、Nの数は、3番染色体を除く全ての染色体でUCSCのとGRCのとが一致しました。
唯一つ、3番染色体は、Nの数が違ったのです。
正確には、
60,830,534番目の塩基が、UCSCではN、GRCではM
60,830,763番目と次の塩基が、UCSCではNN、GRCではRR
でした。
以下、ViewerはCLCのGenomics Workbenchを使用

上段がUCSC hg19、下段がGRCh37の3番染色体
NとMが違います。

こちらはNNとRR
ちなみに、これら塩基のある場所は、FHITという遺伝子のイントロン領域でした。

別に、どうってことは無いんでしょうけど、全く同じだと思っていた予想は外れてしまいました。
30億分の3塩基ですからね。
あと個人的に、Nという塩基がどれくらいあるのか気になったので、染色体ごとにグラフにしてみました。 これはGRCh37を基準にしています。

縦軸は塩基の数です。
全体を揃えて%で見るとこんな感じ。

へえー、Yって半分以上がまだNなんだー。
2011年11月21日月曜日
メチレーションをダイレクトに検出するシーケンサー
シーケンサーは塩基配列を読むもの
というのはSanger型から始まってSolexa型、454型、SOLiD型、Ion Torrent型等、今までのシーケンサーの常識です。
でも、配列だけではなく、メチレーションの有無も同時に読んでくれたら・・・
これがPacBioではできる(らしい)のです。
Flusberg et al. Direct detection of DNA methylation during single-molecule, real-time sequencing.
Nat Methods. 2010 Jun;7(6):461-5.
昨年のthe Advances in Genome Biology and Technology (AGBT)のポスター(実際には行っていないんですけど)から注目していたのですが、PacBioのSingle Molecule Real Time (SMRT)シーケンスを使えば、塩基の修飾も配列と同時に検出することができるようなのです。
SMRTテクノロジーについては以前、
PacBio データの特徴 1 と 2 に書きました。
SMRTからの塩基配列データは、パルス (4種類の異なる光)として出力されます。
 (AGBTポスターより)
(AGBTポスターより)
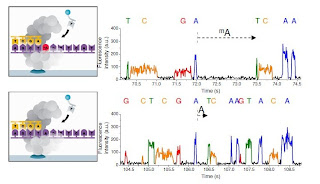
パルスから得られる情報には、塩基そのものの他にも、一塩基が読みとられる時のパルスの幅 Pulse width (PW) と、次の塩基が読みとられるまでのパルスの間隔 Interpulse duration (IPD) もあります。
このIPDが、塩基が修飾されていると変化するのです。
具体的には、Aがメチル化されている N6-methyladenosine (mA) の場合、Tと結合する部分のNにメチル化が起こっているため、Tと結合するのが遅くなるそうです。
そうすると、メチル化されたAのパルスの間隔 IPDが、メチル化されていないAと比べて大きくなります。 (下図の上がメチル化A、下がコントロールA。 メチル化Aの後、次のTまでの間隔が、コントロールのAからTまでの間隔と比べて大きいのがわかります)
この原理を用いて、メチル化されているアデノシン(に限らずシトシンも)を見つけよう! ということです。



というのはSanger型から始まってSolexa型、454型、SOLiD型、Ion Torrent型等、今までのシーケンサーの常識です。
でも、配列だけではなく、メチレーションの有無も同時に読んでくれたら・・・
これがPacBioではできる(らしい)のです。
Flusberg et al. Direct detection of DNA methylation during single-molecule, real-time sequencing.
Nat Methods. 2010 Jun;7(6):461-5.
昨年のthe Advances in Genome Biology and Technology (AGBT)のポスター(実際には行っていないんですけど)から注目していたのですが、PacBioのSingle Molecule Real Time (SMRT)シーケンスを使えば、塩基の修飾も配列と同時に検出することができるようなのです。
SMRTテクノロジーについては以前、
PacBio データの特徴 1 と 2 に書きました。
SMRTからの塩基配列データは、パルス (4種類の異なる光)として出力されます。

パルスから得られる情報には、塩基そのものの他にも、一塩基が読みとられる時のパルスの幅 Pulse width (PW) と、次の塩基が読みとられるまでのパルスの間隔 Interpulse duration (IPD) もあります。
このIPDが、塩基が修飾されていると変化するのです。
具体的には、Aがメチル化されている N6-methyladenosine (mA) の場合、Tと結合する部分のNにメチル化が起こっているため、Tと結合するのが遅くなるそうです。
そうすると、メチル化されたAのパルスの間隔 IPDが、メチル化されていないAと比べて大きくなります。 (下図の上がメチル化A、下がコントロールA。 メチル化Aの後、次のTまでの間隔が、コントロールのAからTまでの間隔と比べて大きいのがわかります)
この原理を用いて、メチル化されているアデノシン(に限らずシトシンも)を見つけよう! ということです。

でもこの検出方法は、うまくいくのでしょうか?
論文ではまず、人工的に作ったメチレーション配列とコントロール配列199塩基を、両方とも何百回も同じところを読んで、平均のメチル配列のIPDと、非メチル配列のIPDを比較しています。
これを見ると、必ずしもメチル化された箇所のIPDが大きく変化するわけではなく、その周辺の塩基のIPDがより影響を受けることがわかりました。

縦軸が塩基ごとのIPDの比(メチルサンプル/ 非メチルサンプル)の平均で、横軸が塩基のポジション、▲印がメチル化の場所です。
上からN6-methyladenosine、5-methylcytosine、5-hydroxymethylcytosine の順で、それぞれN=346、504、393 だそうです。
どうでしょう? メチル化された塩基の場所のIPDが変化しているものもありますが、その前後で大きく変化を示しているパターンのものもあるのがわかります。
N6-methyladenosineでは、メチル化塩基とその5番めの塩基
5-methylcytosineでは、2番、3番、6番めの塩基
5-hydroxymethylcytosineでは、2番、6番めの塩基
のIPDが大きく変化しています。 (縦軸の値が異なるのに注意)
このようなパターンがあることは、メチル化の種類も見分けることができる、ということです。
既存のバイサルファイト法のような形では不可能でしょう。
もちろん何百回も読まなくても、数回、しかし繰り返し同じ所を読むことで、メチル塩基と非メチル塩基を区別できるそうです。

SMRTのCircular topology型DNAテンプレートなら可能なのです。
赤線がメチルAを含むテンプレート、青線がコントロールです。
5回読めばはっきりメチルテンプレートとコントロールの、メチルAのパルスが区別できることを示しています。
人工配列はもういい、という方のために、彼らはDNA adenosine methyltransferase positive (dam+)の大腸菌から取り出したC. elegans fosmid 配列をも読んでいます。
比較対象は、Whole Genome Amplification(WGA)したコントロール配列です。
これは3.7kbの配列にフォーカスして解析しています。

dam+ のIPDをコントロール(WGA)のIPDで割った、IPD Ratio が縦軸です。
Aメチル化はGATC配列に起こりやすいので、non-GATCの塩基(○)とGATC塩基(■)を分けています。
N=106で、平均IPDをプロットしています。
全体的に、non-GATC塩基はIPD ratio が2以下なのに対し、GATC塩基(メチル化塩基)は、IPD Ratioが3から7の間にいるので、区別できる! ということは言えるかな?
ちょっと2500塩基付近の下のGATCが微妙ですが。
実際、これからの技術ですね。
IPDの変化のパターンは、メチル化の種類にだけ依存しているのか、周りの塩基によっても違うのか、生物種によっても違うのか、光学的な条件によっても違ってくるのか。
でも、希望は持てます。 一度パターンがわかってしまえば、シーケンスするだけでメチル化の種類もわかってしまうのですから。
2011年11月10日木曜日
Optical Mapping って
RNA-Seqの続きを書く予定でしたが、ちょっと寄り道して、別なことを。
只今、CBI学会(Chem-Bio Informatics学会)に来ています。
日立ソリューションズのブース&プレゼンで、面白い技術を見つけました。
Optical Mapping というものです。

アメリカでは数年前から導入されていたようですが、このたびマシーンが完成して日本では初めて発表されました。
その名も「全ゲノムソリューション」
外挿遺伝子やリピート領域の同定、ゲノム構造の解析、高精度な菌株同一性に威力を発揮するといいます。
OpGEN 社のテクノロジーは、簡単に言うと全ゲノムレベルで制限酵素のマップを作ることにあります。
測定原理は、
詳しくはこちらのページ
DNAを基板に固定してから制限酵素で切っているので、ばらばらにはならず、Cuttingの位置情報が得られる、というのが重要な点です。
制限酵素のマップを作って、これが次世代シーケンサーの解析にどう役立つかというと、今までのショートリードによるde novo アセンブルは、どうしてもうまく読めないところや、リピートや外からの挿入遺伝子配列などがちゃんとつながらない、という問題がありました。
コンティグはできてもそれがつながらないということです。
それを補正する形で、あらかじめ制限酵素マップという物理的な地図を作り、これをもとにしてアセンブルしたときのコンティグをアライメントしていく、というのがこの技術の良いところです。
Optical Mappingでは、制限酵素のマップはできますが、これには塩基情報はありません。
あるのは一つの制限酵素が切った場所、の情報です。
では、どうやってこの制限酵素マップと、NGSからの配列情報、de novo Assemblyで出来たContigをアラインさせるのでしょうか?
答えは単純でした。
ある一定の長さを持つContigの中の、制限酵素Cutting部位を検索し、Contigひとつひとつに制限酵素マップをつくります。 これは付属のソフトで出来るそうです。
このContig内の制限酵素マップを、Optical Mappingで求めた全ゲノムマップに対してアラインしていくと、Contigに含まれる配列情報が全ゲノムマップに反映される、ことになります。
普通のNGSの感覚では塩基配列を基にアラインすると思ってしまうのですが、これは制限酵素のマップ(Cuttingの場所情報)を基にアラインするのです。 へぇー
実例として、あの大腸菌O-114のゲノムを紹介していました。
PacBio、Ion Torrent、Illumina 等で読まれたデータのContigは、すべてOptical Mappingで作った制限酵素マップがカバーしていました。
現在はバクテリアサイズのゲノムに対応しているそうですが、本国ではヒトゲノムサイズのマップにも成功しているそうです。
最近のモントリオールの人類遺伝学会でも発表したそうですが、普通の(遺伝病などを持たないという意味でしょうが)白人男性の全ゲノムをこの制限酵素マッピング技術で読んで、既知のゲノム配列の制限酵素マップと比較した結果、なんと
こんなに大きな変異が普通にあったとしたら、今のリシークエンスに使っているUCSCのHG19とかの既知ゲノム配列って、何なんだろう、って思いました。
我々が「既知だ」、と思っているヒトゲノムのリファレンス配列が、人によって数メガ塩基の単位で異なるとしたら、 このリファレンス配列にReadをMappingしているだけでは、染色体の構造変化を見つけることはできません。
ドラフト配列だらけの非モデル生物ゲノムについては、言うまでもありません。
改めて、染色体構造を物理的に知ることの重要性を確認しました。
なお、この技術やマシンについては日立ソリューションズさんにお問い合わせください。
きっと親身に説明してくれると思います。
只今、CBI学会(Chem-Bio Informatics学会)に来ています。
日立ソリューションズのブース&プレゼンで、面白い技術を見つけました。
Optical Mapping というものです。

アメリカでは数年前から導入されていたようですが、このたびマシーンが完成して日本では初めて発表されました。
その名も「全ゲノムソリューション」
外挿遺伝子やリピート領域の同定、ゲノム構造の解析、高精度な菌株同一性に威力を発揮するといいます。
OpGEN 社のテクノロジーは、簡単に言うと全ゲノムレベルで制限酵素のマップを作ることにあります。
測定原理は、
- 菌体から長い(200kb以上必要だそうです)ゲノムDNAを抽出し
- ガラス基板上に並べて固定し、
- 制限酵素(1種類)でDNAを切断し
- 蛍光により切断断片の長さを測定
- 制限酵素マップをもとにアセンブルして、全ゲノムの制限酵素マップを作る
詳しくはこちらのページ
DNAを基板に固定してから制限酵素で切っているので、ばらばらにはならず、Cuttingの位置情報が得られる、というのが重要な点です。
制限酵素のマップを作って、これが次世代シーケンサーの解析にどう役立つかというと、今までのショートリードによるde novo アセンブルは、どうしてもうまく読めないところや、リピートや外からの挿入遺伝子配列などがちゃんとつながらない、という問題がありました。
コンティグはできてもそれがつながらないということです。
それを補正する形で、あらかじめ制限酵素マップという物理的な地図を作り、これをもとにしてアセンブルしたときのコンティグをアライメントしていく、というのがこの技術の良いところです。
Optical Mappingでは、制限酵素のマップはできますが、これには塩基情報はありません。
あるのは一つの制限酵素が切った場所、の情報です。
では、どうやってこの制限酵素マップと、NGSからの配列情報、de novo Assemblyで出来たContigをアラインさせるのでしょうか?
答えは単純でした。
ある一定の長さを持つContigの中の、制限酵素Cutting部位を検索し、Contigひとつひとつに制限酵素マップをつくります。 これは付属のソフトで出来るそうです。
このContig内の制限酵素マップを、Optical Mappingで求めた全ゲノムマップに対してアラインしていくと、Contigに含まれる配列情報が全ゲノムマップに反映される、ことになります。
普通のNGSの感覚では塩基配列を基にアラインすると思ってしまうのですが、これは制限酵素のマップ(Cuttingの場所情報)を基にアラインするのです。 へぇー
実例として、あの大腸菌O-114のゲノムを紹介していました。
PacBio、Ion Torrent、Illumina 等で読まれたデータのContigは、すべてOptical Mappingで作った制限酵素マップがカバーしていました。
現在はバクテリアサイズのゲノムに対応しているそうですが、本国ではヒトゲノムサイズのマップにも成功しているそうです。
最近のモントリオールの人類遺伝学会でも発表したそうですが、普通の(遺伝病などを持たないという意味でしょうが)白人男性の全ゲノムをこの制限酵素マッピング技術で読んで、既知のゲノム配列の制限酵素マップと比較した結果、なんと
- 22番長鎖に240kbのInsertion
- 8番短鎖に4.5MbのInversion
こんなに大きな変異が普通にあったとしたら、今のリシークエンスに使っているUCSCのHG19とかの既知ゲノム配列って、何なんだろう、って思いました。
我々が「既知だ」、と思っているヒトゲノムのリファレンス配列が、人によって数メガ塩基の単位で異なるとしたら、 このリファレンス配列にReadをMappingしているだけでは、染色体の構造変化を見つけることはできません。
ドラフト配列だらけの非モデル生物ゲノムについては、言うまでもありません。
改めて、染色体構造を物理的に知ることの重要性を確認しました。
なお、この技術やマシンについては日立ソリューションズさんにお問い合わせください。
きっと親身に説明してくれると思います。
2011年11月4日金曜日
RNA-Seqのカウントって....どうする?
“ There are known knowns; there are things we know we know.
We also know there are known unknowns; that is to say we know there are some things we do not know.
But there are also unknown unknowns -- the ones we don't know we don't know."
We also know there are known unknowns; that is to say we know there are some things we do not know.
But there are also unknown unknowns -- the ones we don't know we don't know."
これは2002年 2月12日、記者から、イラクにあるとされる大量破壊兵器がテロリストの手に渡ったという確かな証拠が無いことを指摘されて、これに答えたドナルド・ラムズフェルド国防長官(当時)の言葉です。
当時、あまりに意味難解な返答に、たくさんのコメディアンがジョークのネタにしていたことを覚えています。
既知なこと、知っていること、わかっていること。 これらが存在することを知っていること、わかっていること。
わからないことがあるということを知っていること。 あるいは知らないこと。
・・・哲学的ですね。
これとRNA-Seqとが何の関係があるかというと、ノーマライズの方法について、知っていることと知っていると仮定していること、知らないこと、をちゃんと知っておく必要があるからです。
タグの数から発現量を算出する時の、RPKMという方法を以前書きました。
RPKMでは、タグカウントを、サンプル内の全カウントで割って、さらに転写産物の長さで割って、定数を乗じて、ノーマライズするのでした。
くわしくはWikiなどを参照。
ここで、
タグ(リード)数はKnownですか?
サンプル内総タグ数はKnownですか?
転写産物の長さはKnownですか?
Isoformの存在、未知Exonの存在などを加味すると、転写産物の長さは、厳密にはUnknownですね。 しかしこれをKnownと仮定して、計算します。
タグ数も、この仮定の転写産物の長さにマップされた数としてカウントされるのですから、厳密にはUnknownです。
こうなってくると、サンプル内総タグ数は、転写産物にMappableなタグという意味では、Knownとするべきでしょうか?
とは言っても、転写産物の長さを厳密に認識するのは、ショートリードでは不可能ですから、みなさんRefSeqなどで公開されている既知のmRNAの長さを用いるのです。
でも頭のどこかに、これは仮定の長さかもしれない、と覚えておくのは意味があるでしょう。
さて、
複数サンプル間で発現量を比較したいとき、遺伝子の長さがとても影響するということがわかっています。
Oshlack et al., Transcript length bias in RNA-seq data confounds systems biology. Biology Direct 2009, 4:14.
転写産物が長い遺伝子ほど、異なるサンプル間で発現量の変動が大きく計算される傾向があるのです。
この論文では遺伝子の長さで補正していません。 発現変動の大きさと、遺伝子の長さをそれぞれ軸にプロットすると、発現変動の大きい遺伝子ほどmRNAが長い遺伝子だということがわかったそうです。

RNA-Seqの「発現変動ありとする」やりかたに問題があるのは、長い遺伝子ほど変動ありとされている遺伝子の割合が高いことから、明らかです。
そこで、RPKMのように転写産物の長さで補正することが必要になるのですが、これまた、RPKMも万能ではありません。
つづく
2011年10月30日日曜日
ExomeとSNVについてのメモ2
Exomeというのは、ゲノム配列からExon、つまり転写領域のみに注目して、その部分の配列のみをシーケンスする方法です。
ゲノム全体を読むよりも、はるかに効率的に、遺伝子領域を高いカバレージで読むことができます。
ゲノムから興味のある配列のみを選択的にセレクトする技術を、ターゲットキャプチャーと呼びますが、いくつかのメーカーからキットが販売されています。
イルミナ社のTrueSeq、ライフテック社のTargetSeq、ロッシュ社のSeqCap、アジレント社のSureSelectがあります。
ここまでは前にも書きました。
では、初めてのひとは、どのキットを使ったら良いのでしょう?
選択にあたっての基準はあるのでしょうか?
当然ながら、アジレント社を除く3社は、自分たちのシーケンサーにもっとも適したプロトコールを用意してキットを設計しているはずです。
しかし、メーカーに問い合わせてみると、「他社のシーケンサー用にも使えますよ」、という答えが返ってきます。 (「でも保証はしませんが」、と念を押されるかもしれませんけど)
考えてみればキャプチャーは、シーケンサーにかけるずっと前の作業なので、キャプチャーに使用した試薬等がきちんとWash outされれば、ライブラリー作成などに問題はおこさないはずです。
問題は、どれだけキャプチャーの効率と、キャプチャーに用いるプローブの設計、何塩基の間隔でプローブが設計されているか、アレイと使うか液体中のビーズを使うか、などによる違いが大きいでしょう。
これらを比較した論文があります。
Performance comparison of exome DNA sequencing technologies.
Nat Biotechnol. 2011 Sep 25;29(10):908-14. PMID:21947028
時間が無いのでサマリーだけ読みたい方はこちら
ヒトのExonキャプチャーキットの比較です。
これによると、プローブの設計にだいぶ特徴があるみたいです。

プローブ長もさることながら、設計場所の「くせ」もあるようです。
Nimblegenはもっとも多くのExon領域を、micro RNA を含め、たくさんカバーしているそうです。
AgilentはExonバリアントの検出に強いそうです。
Illuminaは、UTRの配列もキャプチャーできるそうです。
また、Illuminaはリードデータをもっとも多く必要とするそうです。 最新のHiSeq2000やSOLiD5500ならリードは膨大に出力されるので問題なさそうですが、バーコードを付けて多くのサンプルを読むときは注意が必要ですね。
あくまでヒトExonキャプチャーの比較ですが、バリアントもキャプチャーしたいならAgilent、UTRに注目したいならIllumina、micro RNA も含めてもっとも高いカバレージでキャプチャーしたいならNimblegen、と言えるでしょうか。 かなり大雑把ですが。
私はキャプチャーキットに関しては詳しく知りませんでした。 違いは実験の差くらいに考えていました。
ですが、そのような私にもわかるように、キットの特徴を第三者の目で比較してくれる、こういう論文はうれしいです。
P.S. SNP検出に関するキャプチャーキットの差はあまり無いそうですが、検出されるSNPの差はマッピングやSNP検出ツールのパラメータによるものが大きいと思います。
ゲノム全体を読むよりも、はるかに効率的に、遺伝子領域を高いカバレージで読むことができます。
ゲノムから興味のある配列のみを選択的にセレクトする技術を、ターゲットキャプチャーと呼びますが、いくつかのメーカーからキットが販売されています。
イルミナ社のTrueSeq、ライフテック社のTargetSeq、ロッシュ社のSeqCap、アジレント社のSureSelectがあります。
ここまでは前にも書きました。
では、初めてのひとは、どのキットを使ったら良いのでしょう?
選択にあたっての基準はあるのでしょうか?
当然ながら、アジレント社を除く3社は、自分たちのシーケンサーにもっとも適したプロトコールを用意してキットを設計しているはずです。
しかし、メーカーに問い合わせてみると、「他社のシーケンサー用にも使えますよ」、という答えが返ってきます。 (「でも保証はしませんが」、と念を押されるかもしれませんけど)
考えてみればキャプチャーは、シーケンサーにかけるずっと前の作業なので、キャプチャーに使用した試薬等がきちんとWash outされれば、ライブラリー作成などに問題はおこさないはずです。
問題は、どれだけキャプチャーの効率と、キャプチャーに用いるプローブの設計、何塩基の間隔でプローブが設計されているか、アレイと使うか液体中のビーズを使うか、などによる違いが大きいでしょう。
これらを比較した論文があります。
Performance comparison of exome DNA sequencing technologies.
Nat Biotechnol. 2011 Sep 25;29(10):908-14. PMID:21947028
時間が無いのでサマリーだけ読みたい方はこちら
ヒトのExonキャプチャーキットの比較です。
これによると、プローブの設計にだいぶ特徴があるみたいです。

プローブ長もさることながら、設計場所の「くせ」もあるようです。
Nimblegenはもっとも多くのExon領域を、micro RNA を含め、たくさんカバーしているそうです。
AgilentはExonバリアントの検出に強いそうです。
Illuminaは、UTRの配列もキャプチャーできるそうです。
また、Illuminaはリードデータをもっとも多く必要とするそうです。 最新のHiSeq2000やSOLiD5500ならリードは膨大に出力されるので問題なさそうですが、バーコードを付けて多くのサンプルを読むときは注意が必要ですね。
あくまでヒトExonキャプチャーの比較ですが、バリアントもキャプチャーしたいならAgilent、UTRに注目したいならIllumina、micro RNA も含めてもっとも高いカバレージでキャプチャーしたいならNimblegen、と言えるでしょうか。 かなり大雑把ですが。
私はキャプチャーキットに関しては詳しく知りませんでした。 違いは実験の差くらいに考えていました。
ですが、そのような私にもわかるように、キットの特徴を第三者の目で比較してくれる、こういう論文はうれしいです。
P.S. SNP検出に関するキャプチャーキットの差はあまり無いそうですが、検出されるSNPの差はマッピングやSNP検出ツールのパラメータによるものが大きいと思います。
2011年10月14日金曜日
Exome と SNVについてのメモ 1
最近ブログを更新していませんでした。
先月末から何ていうのでしょう、一種のスランプみたいな、そんな気分でしたので。
もう脱しつつあるので、大丈夫です。
昔からGWASなどでSNPを解析しているひとには常識かもしれませんが、NGSが出た最近やっとSNP(SNV)を始めてみようかな、というひとには意外と「目からうろこ」なことがあります。
私もSNPを昔からやっていたわけではないので、たまに論文を読んでいて気づかされることがあります。
で、そういうことを他の研究者と話すと、意外とその人も知らなかったり。
具体的には、NGSを使ってWhole Exomeをやっている解析で、SNVを見つける、そのときのワークフローです。
当たり前のように行っていたフィルタリングのプロセスで、「なぜそのフィルタリングを行うのか」「基本となる考え方・仮定は何か」という基本的なところがおろそかになっていたと。
そこについてメモ書きのようですが、まとめました。
Exome解析の最終目的は、おおよそ、「疾患原因となるSNVの候補を絞り込む」ことになると思います。
そこで、
それを理解していないでなんとなくパイプライン的に解析を処理していると、後から他の研究者に突っ込まれてタジタジ・・・となってしまう!
以下はSNPをやってたひとには当たり前すぎることを書いていると思います。 どうぞお許しを。

まとめると、上記のワークフローで解析するための前提・仮定は、
「全ての患者に共通するSNVがあって、かつそのSNVを持っていればかなりの確率でその疾患にかかり、そのSNVはとても珍しく、そしてタンパク質のアミノ酸コードを変えることで疾患フェノタイプを引き起こす」
ということになりますでしょうか。
今日は字ばっかりですみませんでした。
先月末から何ていうのでしょう、一種のスランプみたいな、そんな気分でしたので。
もう脱しつつあるので、大丈夫です。
昔からGWASなどでSNPを解析しているひとには常識かもしれませんが、NGSが出た最近やっとSNP(SNV)を始めてみようかな、というひとには意外と「目からうろこ」なことがあります。
私もSNPを昔からやっていたわけではないので、たまに論文を読んでいて気づかされることがあります。
で、そういうことを他の研究者と話すと、意外とその人も知らなかったり。
具体的には、NGSを使ってWhole Exomeをやっている解析で、SNVを見つける、そのときのワークフローです。
当たり前のように行っていたフィルタリングのプロセスで、「なぜそのフィルタリングを行うのか」「基本となる考え方・仮定は何か」という基本的なところがおろそかになっていたと。
そこについてメモ書きのようですが、まとめました。
Exome解析の最終目的は、おおよそ、「疾患原因となるSNVの候補を絞り込む」ことになると思います。
そこで、
- ヒトならヒトのExon領域をカバーしたターゲットシーケンスを行い、その領域で十分な厚みを持ってマップされた場所から、SNVを見つけます。 (例:SureSelectなどでターゲットキャプチャーしたあとIlluminaマシンで大量に読み、BWAでヒトゲノムにマッピング、キャプチャー領域にてSamtoolsでSNVを検出)
- 検出されたSNVの場所とリストをもとに、dbSNPなどのデータベースに無いものを抽出
- さらにその中からNon-synonymousのSNV(アミノ酸置換を伴うSNV)のみを抽出
- そして残ったnsSNVのうち、患者複数のサンプルで共通するものを選び出す
それを理解していないでなんとなくパイプライン的に解析を処理していると、後から他の研究者に突っ込まれてタジタジ・・・となってしまう!
以下はSNPをやってたひとには当たり前すぎることを書いていると思います。 どうぞお許しを。
- Exon領域をキャプチャーする大前提は、テーマとしている疾患の変異を、遺伝子のコーディング領域から探そうとしているわけです。 つまりコーディングされない場所にいくら疾患原因SNVがあったとしても、それは見ないことにしよう、としています。
もちろんコーディング領域は重要です。 しかしそれ以外の転写制御領域のSNVも大変重要ですが、これらは一般的なExomeでは解析対象から外されます。 - dbSNPに無い、SNVのみを探すというのは、一見、新規性を探索するのに合理的な方法です。 この疾患原因SNVは非常にレア、珍しいという前提があります。 しかし、データベースに登録されるSNVは、現在爆発的に増え続けていますので、既知であっても自分の研究疾患では新規ということは十分あり得ます。
- Non-synonymousのSNVに絞るということは、アミノ酸置換、あるいはフレームシフト、ミッセンス等を伴うような変異は特に重要だという前提に立っています。 「この疾患の原因は、アミノ酸変異を伴うSNVだ」 という仮定がそこにあります。 そうでない仮定の場合にNon-synonymous SNVに絞るのは適当でないでしょう。
また気をつけるべきは、Alternative Exon(スプライスバリアント)の存在です。 これを無視してSNVを見つけているケースが多いのではないでしょうか。 ゲノムのSNVではなく遺伝子コーディング領域中のSNVとなると、スプライスバリアントが異なればそのSNVの有る無しの意味は大きいはずですよね。 (言葉足らず) - 複数の患者のサンプルで同じSNVを見つけるというのは、そのフェノタイプに共通するSNVを見つけ出すということです。 SNV(原因)があれば必ずそのフェノタイプ(結果)を引き起こすことをComplete Penetranceと言います。 一方、そのフェノタイプ(結果)には必ずそのSNV(原因)が見られるときをComplete Detectance と言います。
つまりこのフィルタリングは、全ての患者に共通するSNVがあるはずだ、という前提に立っているわけです。
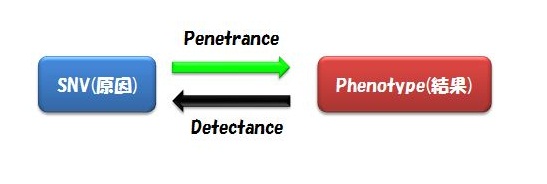
まとめると、上記のワークフローで解析するための前提・仮定は、
「全ての患者に共通するSNVがあって、かつそのSNVを持っていればかなりの確率でその疾患にかかり、そのSNVはとても珍しく、そしてタンパク質のアミノ酸コードを変えることで疾患フェノタイプを引き起こす」
ということになりますでしょうか。
今日は字ばっかりですみませんでした。
2011年9月27日火曜日
PacBio データの特徴 2
PacBioの実験ワークフローで、とてもユニークなのがテンプレートの形です。
ダンベル型のSMRTBellというそうです。
Travers et al., A flexible and efficient template format for circular consensus sequencing and SNP detection (2010). Nucleic Acids Research 38, e159.
 DNAをフラグメント化して、断片の両端をエンドリペア後、輪っか配列(プライマーのため)を付けてAのダンベル型テンプレートができます。
DNAをフラグメント化して、断片の両端をエンドリペア後、輪っか配列(プライマーのため)を付けてAのダンベル型テンプレートができます。
左端の、輪っかの部分がシングル鎖になっていますね。ここにプライマーがくっつきます。
ポリメラーゼは、DNAの二重鎖のところを押しのけて、プライマーの先に、相補鎖に正しい塩基を合成していきます。
テンプレートは、ずーっといくと一周しますね。
そうしたらまた、Bのように、今合成した通りにもう一度合成をするのです。
つまり、インサートの長さに依存することなく、何度でもシーケンスが可能です。
これがダンベル型テンプレート。
このままの形がスタンダードです。
インサートの長さがものすごーく長い(数10kbとか)とき、全部読むことはできないので輪っかの近いところだけを読むことになります。 これはMait-Pairのような感じです。
Strobeと呼ぶそうです。
もうひとつはインサートの長さは適度で、テンプレートを何周も読む方法。
何度も何度も同じ配列を読むことになるので、精度は上がります。
Circular Consensusと呼ぶそうです。
ここから先は、スタンダードのテンプレートの話です。
SMRTでは、インサートを全部読むので、Read長=インサート長、になります。(SMRTについては前回のブログを参照)
しかし、全部読み切れなかったインサートもあるでしょう。
そんな、「途中まで読めたインサート」配列が混じっている状態のReadのことを、Subreadと呼ぶそうです。
Subreadがいくつか集まってRead(=インサート配列)を形成する、ということです。
ですから、SMRTで読んだ解析結果には必ず、
Subread の平均の長さ
Read の平均の長さ、95パーセンタイルの長さ、・・・
など、Subread とReadの両方の情報があるのが普通でしょう。
前回紹介した、Expression Analysis社のWebセミナーで紹介していたのは、SMRTで大腸菌のゲノムを読んだときのデータ解析についてです。
深夜2時過ぎのことでしたので私の記憶が正しく無い箇所もあるかもしれません。
2kbと6kbの2種類のインサートを読んでいました。
SMRTのひとつのセル(フローセルのような単位)で、34~108 MbのMappableなデータが出力されました。
平均1500塩基長、プロトコルを改良すると2700塩基長を一度に読めていました。
特徴のひとつは、大腸菌ゲノムに対するカバレージが一定だったことです。
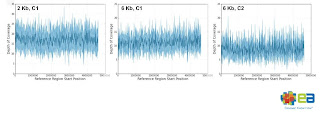 普通、GC含量のばらつきなどにより、読みにくかったりする箇所がどうしても出てくるものですが、
普通、GC含量のばらつきなどにより、読みにくかったりする箇所がどうしても出てくるものですが、
それが比較的無くカバレージが一定で、
2kbインサートが6.4~17.1カバレージ
6kbインサートが6.7~11.2カバレージをゲノム全体で保持していたそうです。
精度ですが、Subreadのレベルの精度は85%程だったそうです。
これがSMRTの精度だと一般的に言われると、15%は間違い? 結構高いね、と思われそうですが、SubreadではなくRead、あるいは何度も繰り返して読んだ時のConsensusともなると、96.46~99.39%まで向上していたそうです。
何度も読むというのは、賢い方法です。
ダンベル型テンプレートならでは?
今はまだ、バクテリアサイズのゲノムシーケンスに用いられているPacBioのSMRTです。
ランニングコストがどれくらいなのかはわかりませんが、もっと汎用的に使われる時はもうすぐ来るのでしょうか。
個人的には、機械が大きすぎ! って思います。

ダンベル型のSMRTBellというそうです。
Travers et al., A flexible and efficient template format for circular consensus sequencing and SNP detection (2010). Nucleic Acids Research 38, e159.

左端の、輪っかの部分がシングル鎖になっていますね。ここにプライマーがくっつきます。
ポリメラーゼは、DNAの二重鎖のところを押しのけて、プライマーの先に、相補鎖に正しい塩基を合成していきます。
テンプレートは、ずーっといくと一周しますね。
そうしたらまた、Bのように、今合成した通りにもう一度合成をするのです。
つまり、インサートの長さに依存することなく、何度でもシーケンスが可能です。
これがダンベル型テンプレート。
このままの形がスタンダードです。
インサートの長さがものすごーく長い(数10kbとか)とき、全部読むことはできないので輪っかの近いところだけを読むことになります。 これはMait-Pairのような感じです。
Strobeと呼ぶそうです。
もうひとつはインサートの長さは適度で、テンプレートを何周も読む方法。
何度も何度も同じ配列を読むことになるので、精度は上がります。
Circular Consensusと呼ぶそうです。
ここから先は、スタンダードのテンプレートの話です。
SMRTでは、インサートを全部読むので、Read長=インサート長、になります。(SMRTについては前回のブログを参照)
しかし、全部読み切れなかったインサートもあるでしょう。
そんな、「途中まで読めたインサート」配列が混じっている状態のReadのことを、Subreadと呼ぶそうです。
Subreadがいくつか集まってRead(=インサート配列)を形成する、ということです。
ですから、SMRTで読んだ解析結果には必ず、
Subread の平均の長さ
Read の平均の長さ、95パーセンタイルの長さ、・・・
など、Subread とReadの両方の情報があるのが普通でしょう。
前回紹介した、Expression Analysis社のWebセミナーで紹介していたのは、SMRTで大腸菌のゲノムを読んだときのデータ解析についてです。
深夜2時過ぎのことでしたので私の記憶が正しく無い箇所もあるかもしれません。
2kbと6kbの2種類のインサートを読んでいました。
SMRTのひとつのセル(フローセルのような単位)で、34~108 MbのMappableなデータが出力されました。
平均1500塩基長、プロトコルを改良すると2700塩基長を一度に読めていました。
特徴のひとつは、大腸菌ゲノムに対するカバレージが一定だったことです。
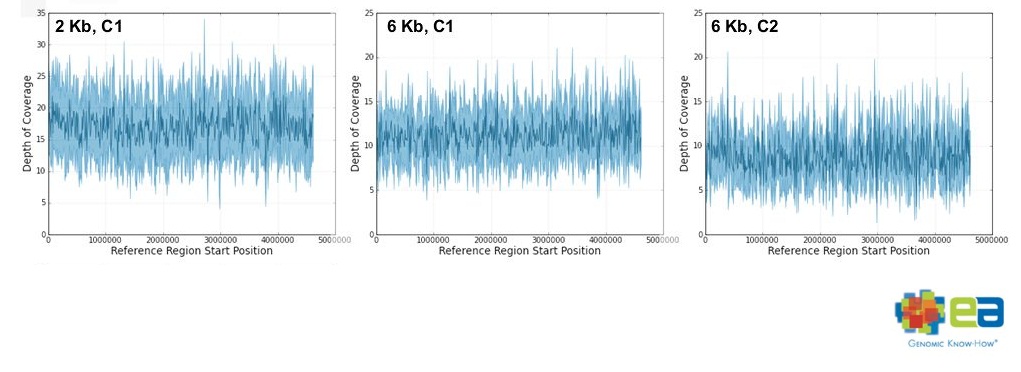
それが比較的無くカバレージが一定で、
2kbインサートが6.4~17.1カバレージ
6kbインサートが6.7~11.2カバレージをゲノム全体で保持していたそうです。
精度ですが、Subreadのレベルの精度は85%程だったそうです。
これがSMRTの精度だと一般的に言われると、15%は間違い? 結構高いね、と思われそうですが、SubreadではなくRead、あるいは何度も繰り返して読んだ時のConsensusともなると、96.46~99.39%まで向上していたそうです。
何度も読むというのは、賢い方法です。
ダンベル型テンプレートならでは?
ランニングコストがどれくらいなのかはわかりませんが、もっと汎用的に使われる時はもうすぐ来るのでしょうか。
個人的には、機械が大きすぎ! って思います。

もうちょい、コンパクトにならないのか・・・
2011年9月23日金曜日
PacBio データの特徴 1
PacBioのシーケンサーから出てくるデータはどんなものなんだろう?
その前に、「4分でわかるPacBio-SMRTテクノロジー」をYouTubeで見つけたので、SMRTって何だ?ってひとはまずはここから。
テクノロジーについてもっと詳しく知りたい方は、PacBioのホームページか、この論文がお勧めです。
Schadt et. al., "A window into third-generation sequencing". Hum. Mol. Genet. (2010) 19 (R2): R227-R240
SMRTの特徴は、DNAを1分子単位で、PCRによる増幅無しで、結構長く読めるということです。
Single Molecule Real Time の略です。
ガラススライド上に無数の小さなポケット(ZMWs (zero-mode waveguides) )があり、その穴の底には、DNAポリメラーゼが固定されています。 この穴は非常に小さく、波長600nmまでの可視光線は通り抜けできない構造になっています。
シーケンスは、プライマーがアニールされたDNA鋳型がポリメラーゼに取り込まれ、反応準備が完了します。
4種類の異なる蛍光の付いた塩基が、ZMWsの外から入り込み、DNA鋳型に取り込まれると蛍光が切り離されてZMWsの穴の底で光ります。その光は底から30ナノメートルまでしか届きません。
スキャナーはその光を検出します。 底にはポリメラーゼが固定されているので、その近くで観測される光は、取り込まれた塩基による光しかない、というわけです。
でもランダムにZMWsの外からも蛍光付きの塩基が入り込むので、偶然ポリメラーゼ近辺に来て、蛍光が検出されてしまうことはないのでしょうか。
IlluminaやSOLiDのような、蛍光検出一回につき余分な蛍光を洗い流す、Wash-and-Detectはしていません。
あくまでもReal-Timeにポリメラーゼが自然に塩基がとりこまれるままで読んでいるのです。
トリックは、ポリメラーゼに塩基がとりこまれる速度はミリ秒単位、それ以外のZMWs内の塩基の出入りはマイクロ秒単位、という時間の差にあります。
ミリ秒単位で光らないと、DNA鎖の塩基として認識しないのです。

先日、Expression Analysis(http://www.expressionanalysis.com/)というアメリカの会社がPacBioのデータ解析Webセミナーをやっていたので、深夜2時だったけど参加してみました。
PacBioのデータって、どんなんだろう? すごく興味があったせいか、3時までずっと起きていました。 次の日会社・・・
さて、SMRTテクノロジーで検出される塩基は、ポリメラーゼがReal Timeに合成する速度と関係が深くなります。
ということは、塩基の検出される時間幅は、一定では無いということです。
Webセミナーの中でもそれを言っていました。

とは言っても、PacBioの機械・ソフトは、これを何とか解決しているのでしょう。 でなければ商品化していませんよね。
次は、PacBioのデータを解釈するにあたって絶対必要な、リード、サブリード、ダンベル型のSMRTBellテンプレート、について説明します。
2011年9月10日土曜日
NGSでmicroRNA 2
前回はNGSでmicroRNAを読むときの一般的なワークフローで、マッピングまで書きました。
既知のmiRNA配列にマップしたときは、その発現量がマップされたリードの本数で表現できる、ということは直感的にわかるでしょう。
以前、RNA-Seqの発現量推定のところでも書きましたが、リードの数はサンプルごとのノーマライズ(正規化)が必要です。
(その既知miRNAにマップされたリードの本数)/ (そのサンプルランで取れた全リードのうちmiRNAにマップされたリードの合計数)
出力されたリードの数、特にマッパブル(Mappabl)なリードの総数は、サンプルによって異なります。 ですから、各miRNAにマップされたリードの本数を全体で割ってあげて、異なるサンプル間で比較可能にするのです。
分母を、そのサンプルのリード本数全体 として計算している方もおられますが、私は、マップされたリード本数全体 とした方が精度が上がると考えています。
だってマップされないリードまでノーマライズの分母に含めるのは不自然でしょう。
これだけでは値が非常に小さくなるので、100万倍して、RPM(Read Per Million)という表現にするのが良いかも知れません。
さて、そうして正規化された値は、当然ながらmiRNAの発現量そのものではありません。
あくまで比です。
ということで、この値は、ほかのサンプルの正規化miRNA発現値と比較して、何倍発現量が多い、少ない、というような解析に持っていきます。
マイクロアレイと同じですね。
Nが3以上あれば、正規化後の値を対数変換してから、通常の統計手法(T‐Testなど)で比較すると良いでしょう。
DESeqという発現量比較のツールも使えるかと思います。
いろいろ調べてみると結構miRNA関連論文で、このDESeqが使われています。
元々は発現変動遺伝子を解析するツールです。
遺伝子は全部で数万あり、その中でほとんどは変動していない、という仮定の下、変動有意遺伝子を見つけるわけです。
microRNAは全部でもたかだか数百、これで遺伝子発現と同じモデル(負の二項分布)式を使っても良いのか、という疑問は残りますが、ここではそれを置いておくとして。
Nが1しか無い場合、割り算して比較するしかありませんね。
有意な発現変動を示したmiRNAが見つかったら、そのターゲット遺伝子を探してみましょうか。
データベースとしてはまず、miRBase (http://www.mirbase.org)がいいでしょう。
 ここからはTargetScan や Pictar といった、ターゲット予測プログラムのサイトへもリンクできます。
ここからはTargetScan や Pictar といった、ターゲット予測プログラムのサイトへもリンクできます。
ターゲット遺伝子を見つけたら、その遺伝子のGO(Gene Ontology)を検索して、どんな機能の遺伝子がターゲットになっていたのか、を調べることも良く行われます。
さて、これら以外に、NGSならではの解析とは何でしょう?
そう、既知ではなく、未知のmiRNAを見つけることです。
それには先ず、マッピングのデータから、既知miRNA以外の場所にマップしたリードが作っている、ある程度の長さのクラスターを見つけてこなければいけません。
ある程度の長さって、どれくらいだ?
クラスター配列は、2次構造を取り得る、いわゆるpalindromicの配列でなくてはなりません。
1次元で言うと、(mature miRNA配列)+(ループ構造)+(mature* miRNA配列)のパターンである必要があります。
そのような配列を取ってきたら、エネルギーが最小になるようなときにちゃんとヘアピンのようにフォールドするかどうか、を確かめます。
さらに、Droshaなどのタンパク質に切断されるサイトがあるか、調べます。
これを全部プログラムを作って行うのは大変ですが、幸い、アカデミックの方ならフリーで使えるツールがあります。 miRDeep というものです。
 このドラえもんの手、みたいなのがmature miRNA-Loop-star の2次構造で、薄い線がマップされているリードです。
このドラえもんの手、みたいなのがmature miRNA-Loop-star の2次構造で、薄い線がマップされているリードです。
matureの方により多くのリードがマップされているのは、より本物らしいmicroRNAだったことを裏付けます。
残念ながら、企業はフリーじゃないんですよね。 いくらか払うのかな。
2次構造だけの予測なら、できるソフトもありますが、miRDeep ほど特化しているものは少ないでしょう。 チャンスがあれば使ってみたいです。
microRNAをNGSで解析するというテーマで、よくまとめられているレビューがあるので紹介します。
Motameny et al. (2010) Next Generation Sequencing of miRNAs - Strategies, Resources and Methods. Genes. 1, 70.
こちらはmiRanalyzerというツールのワークフローです。フローは参考にしています。
Stark et al. (2010) Characterization of the Melanoma miRNAome by Deep Sequencing. PLoS One. 5, e9685.
発現量比較から新規miRNA予測、ターゲットのGO解析まで行った例です。 こちらも良い。
Dhahbi et al. (2011) Deep Sequencing Reveals Novel MicroRNAs and Regulation of MicroRNA Expression during Cell Senescence. PLoSOne. 6, e20509.
既知のmiRNA配列にマップしたときは、その発現量がマップされたリードの本数で表現できる、ということは直感的にわかるでしょう。
以前、RNA-Seqの発現量推定のところでも書きましたが、リードの数はサンプルごとのノーマライズ(正規化)が必要です。
(その既知miRNAにマップされたリードの本数)/ (そのサンプルランで取れた全リードのうちmiRNAにマップされたリードの合計数)
出力されたリードの数、特にマッパブル(Mappabl)なリードの総数は、サンプルによって異なります。 ですから、各miRNAにマップされたリードの本数を全体で割ってあげて、異なるサンプル間で比較可能にするのです。
分母を、そのサンプルのリード本数全体 として計算している方もおられますが、私は、マップされたリード本数全体 とした方が精度が上がると考えています。
だってマップされないリードまでノーマライズの分母に含めるのは不自然でしょう。
これだけでは値が非常に小さくなるので、100万倍して、RPM(Read Per Million)という表現にするのが良いかも知れません。
さて、そうして正規化された値は、当然ながらmiRNAの発現量そのものではありません。
あくまで比です。
ということで、この値は、ほかのサンプルの正規化miRNA発現値と比較して、何倍発現量が多い、少ない、というような解析に持っていきます。
マイクロアレイと同じですね。
Nが3以上あれば、正規化後の値を対数変換してから、通常の統計手法(T‐Testなど)で比較すると良いでしょう。
DESeqという発現量比較のツールも使えるかと思います。
いろいろ調べてみると結構miRNA関連論文で、このDESeqが使われています。
元々は発現変動遺伝子を解析するツールです。
遺伝子は全部で数万あり、その中でほとんどは変動していない、という仮定の下、変動有意遺伝子を見つけるわけです。
microRNAは全部でもたかだか数百、これで遺伝子発現と同じモデル(負の二項分布)式を使っても良いのか、という疑問は残りますが、ここではそれを置いておくとして。
Nが1しか無い場合、割り算して比較するしかありませんね。
有意な発現変動を示したmiRNAが見つかったら、そのターゲット遺伝子を探してみましょうか。
データベースとしてはまず、miRBase (http://www.mirbase.org)がいいでしょう。

ターゲット遺伝子を見つけたら、その遺伝子のGO(Gene Ontology)を検索して、どんな機能の遺伝子がターゲットになっていたのか、を調べることも良く行われます。
さて、これら以外に、NGSならではの解析とは何でしょう?
そう、既知ではなく、未知のmiRNAを見つけることです。
それには先ず、マッピングのデータから、既知miRNA以外の場所にマップしたリードが作っている、ある程度の長さのクラスターを見つけてこなければいけません。
ある程度の長さって、どれくらいだ?
クラスター配列は、2次構造を取り得る、いわゆるpalindromicの配列でなくてはなりません。
1次元で言うと、(mature miRNA配列)+(ループ構造)+(mature* miRNA配列)のパターンである必要があります。
そのような配列を取ってきたら、エネルギーが最小になるようなときにちゃんとヘアピンのようにフォールドするかどうか、を確かめます。
さらに、Droshaなどのタンパク質に切断されるサイトがあるか、調べます。
これを全部プログラムを作って行うのは大変ですが、幸い、アカデミックの方ならフリーで使えるツールがあります。 miRDeep というものです。


matureの方により多くのリードがマップされているのは、より本物らしいmicroRNAだったことを裏付けます。
残念ながら、企業はフリーじゃないんですよね。 いくらか払うのかな。
2次構造だけの予測なら、できるソフトもありますが、miRDeep ほど特化しているものは少ないでしょう。 チャンスがあれば使ってみたいです。
microRNAをNGSで解析するというテーマで、よくまとめられているレビューがあるので紹介します。
Motameny et al. (2010) Next Generation Sequencing of miRNAs - Strategies, Resources and Methods. Genes. 1, 70.
こちらはmiRanalyzerというツールのワークフローです。フローは参考にしています。
Stark et al. (2010) Characterization of the Melanoma miRNAome by Deep Sequencing. PLoS One. 5, e9685.
発現量比較から新規miRNA予測、ターゲットのGO解析まで行った例です。 こちらも良い。
Dhahbi et al. (2011) Deep Sequencing Reveals Novel MicroRNAs and Regulation of MicroRNA Expression during Cell Senescence. PLoSOne. 6, e20509.
2011年9月5日月曜日
NGSでmicroRNA解析 1
microRNAというのは、タンパク質にはならない、いわゆる non-coding RNAの仲間です。
それ自身で標的のmRNAの3’-UTRなどに結合し、標的mRNAを分解します。
microRNAは発現制御機能を持った、RNA分子なのです。
と、ごくごく簡潔にまとめましたが、microRNAについては、多くのレビューや教科書に記載がありますので、詳しくは書かないとして。 Wikipediaでもかなり情報が得られますよ。

さて、microRNAの解析は、長らく、TaqManアレイやその他のマイクロアレイ、PCRなどが主流でした。
近年、NGSでもこれを解析している例が見られます。
実験プロトコールとして一般的なのは、
短めに読む、というのはフラグメントの長さが 18-30塩基を予想しているからで、35、6塩基も読めば十分全体をカバーするからです。
ゲノムや転写産物を読む場合は、できるだけ長く読めたほうが良いですが、microRNAは短くてもOK、むしろちゃんと正確に読めることが大切です。
さて、シーケンサーが無事データを出力しました。
ここからデータ解析です。
microRNAの解析では、リードのアダプタートリミングは大切です。 (microRNAに限ったことではありませんが)
読んでいるフラグメントの長さが短い分、5’側のアダプターから読んだ時に、フラグメントを読み切り、3’側のアダプターまで読んでしまうことがあるからです。
そのため、3’側のアダプターをトリミング(除去)することが必須です。
次に、トリミングした後のリードが、短くなりすぎると、後でマッピングする際にNon Specificにマップされる恐れがあるので、これも除きます。 15‐18塩基以下の短いリードは除去するといいかもしれません。
リードがきれいになったところで、いよいよマッピングです。 でも何に対して?
一例を示します。 今度参考文献も示しますね。
これにより、①既知のmicroRNA、②それ以外のnon-coding RNA、③新規microRNA 候補、の3種類のデータセットができる。
既知のmicroRNA、non-coding RNA については、発現量の解析をすることになるでしょう。
新規候補の解析は、少しやっかいだが、アルゴリズムが無くはないのです。
つづく
それ自身で標的のmRNAの3’-UTRなどに結合し、標的mRNAを分解します。
microRNAは発現制御機能を持った、RNA分子なのです。
と、ごくごく簡潔にまとめましたが、microRNAについては、多くのレビューや教科書に記載がありますので、詳しくは書かないとして。 Wikipediaでもかなり情報が得られますよ。

さて、microRNAの解析は、長らく、TaqManアレイやその他のマイクロアレイ、PCRなどが主流でした。
近年、NGSでもこれを解析している例が見られます。
実験プロトコールとして一般的なのは、
- トータルRNAを抽出して
- ゲルに流した後18-30塩基のあたりを切り出し
- Small RNAを精製して
- ライブラリーキットにある通りアダプターを付け
- cDNA合成し
- ライブラリーをシーケンサーで短めに読む
短めに読む、というのはフラグメントの長さが 18-30塩基を予想しているからで、35、6塩基も読めば十分全体をカバーするからです。
ゲノムや転写産物を読む場合は、できるだけ長く読めたほうが良いですが、microRNAは短くてもOK、むしろちゃんと正確に読めることが大切です。
さて、シーケンサーが無事データを出力しました。
ここからデータ解析です。
microRNAの解析では、リードのアダプタートリミングは大切です。 (microRNAに限ったことではありませんが)
読んでいるフラグメントの長さが短い分、5’側のアダプターから読んだ時に、フラグメントを読み切り、3’側のアダプターまで読んでしまうことがあるからです。
そのため、3’側のアダプターをトリミング(除去)することが必須です。
次に、トリミングした後のリードが、短くなりすぎると、後でマッピングする際にNon Specificにマップされる恐れがあるので、これも除きます。 15‐18塩基以下の短いリードは除去するといいかもしれません。
リードがきれいになったところで、いよいよマッピングです。 でも何に対して?
一例を示します。 今度参考文献も示しますね。
- 先ずゲノムにマッピングし、マップされなかった配列はごみとして除去する
- ゲノムに当たったリードを回収、miRBase の配列(precursor, mature, mature*)にマッピング
- miRBaseの配列にぴったり当たったリードは、既知のmicroRNAとして保存
- miRBaseの配列に当たらなかったリードは、次に、microRNA以外の、既知のnon-coding RNA (piwiRNA, snRNA, snoRNAなどの) 配列に対してマッピング
- non-coding RNAにも当たらなかったリードは、念のため、RefSeqのmRNA配列に対してマッピング
- それでも当たらなかったリードは、新規microRNA「候補」として保存
これにより、①既知のmicroRNA、②それ以外のnon-coding RNA、③新規microRNA 候補、の3種類のデータセットができる。
既知のmicroRNA、non-coding RNA については、発現量の解析をすることになるでしょう。
新規候補の解析は、少しやっかいだが、アルゴリズムが無くはないのです。
つづく
2011年8月27日土曜日
SIFT SNP機能予測アルゴリズム
今日もSNVの話です。
1塩基の変異が、タンパク質にどんな影響を与え得るのか、の予測アルゴリズムです。
今までPolyPhenとかGrantham Scoreとかについて少し触れましたが、今日はSIFTというアルゴリズム&ツールについて。
"SIFT" ってググると、上位には画像検出アルゴリズムがたくさん出てきますが、これは違うので必ず、"SIFT SNP" とかで検索して下さいね。 (画像検出の方も興味がありますが)
http://sift.jcvi.org/
 SIFTとは、Sorting Intolerant From Tolerant の略です。
SIFTとは、Sorting Intolerant From Tolerant の略です。
???
そもそもtolerant とは何でしょう?
辞書で引くと寛容な、とか耐性、とかがあります。
ここではタンパク質の機能に変化が無い、ということを意味します。
SIFTの考え方の基本は、配列保存性です。
同じタンパクファミリーの中で、保存性の高いアミノ酸配列があったとします。
そのような配列の中のアミノ酸変異・置換は、タンパク質の機能に致命的な影響を与える可能性が高いと思います。
機能を失ったタンパク質は、進化の過程で生き残れませんね。
反対に、タンパク質機能に重要な影響を与えない部分のアミノ酸ならば、進化の途中でどんどん置換が起こり得るだろうし、またそのような機能に影響の無い場所のアミノ酸置換は、生き残っているタンパク質にも見られるはずでしょう。
つまり、配列の保存性が高い場所のアミノ酸置換は、タンパク質機能の変化という点でIntolerant(影響あり)。
配列保存性の低い場所のアミノ酸置換は、タンパク質機能変化にTolerant(影響なし)。
この方法は、正しいタンパク質ファミリー(オーソログタンパク)を集めてアライメントすることが重要です。その中で配列保存性を見ていくからです。
UniprotやNCBIのnrなどから配列データを借りています。
さて、このように配列相同性のみを使用した予測ツールでは、タンパク質の立体構造までは見ていません。
ループ構造部分に変異があるのか、疎水性が失われるのか、といった情報までは見ていないのです。
これらは別の予測ツール(Polyphenなど)が必要です。
NGSでSNVを見つけた。
Non-synonymousのSNVを絞り出した。
次に考えるのは、そのnsSNVの重要性です。
そのアミノ酸が変わったらタンパク質として生き残れないよ、という情報は、重要性の指標になります。
そのアミノ酸が変わっても、タンパク質は生き残っていたのなら、その置換はさほど重要ではないでしょう。
最初にSIFTなどで、配列保存性を基準にnsSNVの重要性をフィルターにかけ、残った重要そうなやつを、次にタンパク質立体構造上で確認していく方が近道のような気がします。
さて、実際にNGSでSNVを見つけてからこのツールを使って解析するにはどうしたら良いか?
先にURLを示した場所よりも、以下から入る方がお勧めです。
VCFフォーマットをSIFT専用のフォーマットにコンバートしてくれるからです。
http://sift.bii.a-star.edu.sg/www/SIFT_intersect_coding_submit.html
 ここで、SAMtoolsなどで求めたVCFファイルをアップします。
ここで、SAMtoolsなどで求めたVCFファイルをアップします。
そうするとしばらくして、コンバートされたファイルができますので、これをPCに保存します。
SNP用のファイルと、InDel用のファイルの2種類できます。
ここではSNP用ファイルを例に、落とします。
 保存したら、upload here のリンクをクリックして、そのファイルをアップロードします。
保存したら、upload here のリンクをクリックして、そのファイルをアップロードします。
その時、自分のE-mail アドレスと、オプションでどんなアノテーションを付けたいかを指定します。
 結果は、HTML、またはテキストで参照できます。
結果は、HTML、またはテキストで参照できます。
これがSNPの結果

 リンクできるところはそれぞれのDBにリンクしています。
リンクできるところはそれぞれのDBにリンクしています。
HTMLだと、ちょっと扱いにくいので、私はテキスト(tsv)で落として、Excelで見ることもお勧めします。
例えば、こんな風に絞り込めば、新規っぽいSNPの機能を予測できます。

この絞り込みの条件は、
その時、ScoreとMedian Infoにも注目します。
Scoreは0から1の値を取り、0.05以下のとき、タンパク質機能に影響あり(=Intolerant = Damaging)とされます。
Median Infoは保存性の信頼度で、0から4.32の値をとり、大きいほど信頼度が低くなります。 3.25以上のときはLow confidenceとして警告が出ます。
どうでしょう? SNPの機能を調べるにはとても使いやすいツールだと思います。
もっと詳しく知りたい方は、
http://sift.jcvi.org/www/SIFT_help.html
Kumar P et al. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature Protocols 4, 1073-82 (2009).
1塩基の変異が、タンパク質にどんな影響を与え得るのか、の予測アルゴリズムです。
今までPolyPhenとかGrantham Scoreとかについて少し触れましたが、今日はSIFTというアルゴリズム&ツールについて。
http://sift.jcvi.org/

???
そもそもtolerant とは何でしょう?
辞書で引くと寛容な、とか耐性、とかがあります。
ここではタンパク質の機能に変化が無い、ということを意味します。
SIFTの考え方の基本は、配列保存性です。
同じタンパクファミリーの中で、保存性の高いアミノ酸配列があったとします。
そのような配列の中のアミノ酸変異・置換は、タンパク質の機能に致命的な影響を与える可能性が高いと思います。
機能を失ったタンパク質は、進化の過程で生き残れませんね。
反対に、タンパク質機能に重要な影響を与えない部分のアミノ酸ならば、進化の途中でどんどん置換が起こり得るだろうし、またそのような機能に影響の無い場所のアミノ酸置換は、生き残っているタンパク質にも見られるはずでしょう。
つまり、配列の保存性が高い場所のアミノ酸置換は、タンパク質機能の変化という点でIntolerant(影響あり)。
配列保存性の低い場所のアミノ酸置換は、タンパク質機能変化にTolerant(影響なし)。
この方法は、正しいタンパク質ファミリー(オーソログタンパク)を集めてアライメントすることが重要です。その中で配列保存性を見ていくからです。
UniprotやNCBIのnrなどから配列データを借りています。
さて、このように配列相同性のみを使用した予測ツールでは、タンパク質の立体構造までは見ていません。
ループ構造部分に変異があるのか、疎水性が失われるのか、といった情報までは見ていないのです。
これらは別の予測ツール(Polyphenなど)が必要です。
NGSでSNVを見つけた。
Non-synonymousのSNVを絞り出した。
次に考えるのは、そのnsSNVの重要性です。
そのアミノ酸が変わったらタンパク質として生き残れないよ、という情報は、重要性の指標になります。
そのアミノ酸が変わっても、タンパク質は生き残っていたのなら、その置換はさほど重要ではないでしょう。
最初にSIFTなどで、配列保存性を基準にnsSNVの重要性をフィルターにかけ、残った重要そうなやつを、次にタンパク質立体構造上で確認していく方が近道のような気がします。
さて、実際にNGSでSNVを見つけてからこのツールを使って解析するにはどうしたら良いか?
先にURLを示した場所よりも、以下から入る方がお勧めです。
VCFフォーマットをSIFT専用のフォーマットにコンバートしてくれるからです。
http://sift.bii.a-star.edu.sg/www/SIFT_intersect_coding_submit.html

そうするとしばらくして、コンバートされたファイルができますので、これをPCに保存します。
SNP用のファイルと、InDel用のファイルの2種類できます。
ここではSNP用ファイルを例に、落とします。

その時、自分のE-mail アドレスと、オプションでどんなアノテーションを付けたいかを指定します。

これがSNPの結果


HTMLだと、ちょっと扱いにくいので、私はテキスト(tsv)で落として、Excelで見ることもお勧めします。
例えば、こんな風に絞り込めば、新規っぽいSNPの機能を予測できます。

- dbSNPに登録の無い "novel" SNPで
- アミノ酸置換をもたらす Non-synonymous SNP
その時、ScoreとMedian Infoにも注目します。
Scoreは0から1の値を取り、0.05以下のとき、タンパク質機能に影響あり(=Intolerant = Damaging)とされます。
Median Infoは保存性の信頼度で、0から4.32の値をとり、大きいほど信頼度が低くなります。 3.25以上のときはLow confidenceとして警告が出ます。
どうでしょう? SNPの機能を調べるにはとても使いやすいツールだと思います。
もっと詳しく知りたい方は、
http://sift.jcvi.org/www/SIFT_help.html
Kumar P et al. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature Protocols 4, 1073-82 (2009).
2011年8月20日土曜日
Ensembl - Variant Effect Predictor でSNPアノテーション
先日、VCFフォーマットを読みこんでSNPのアノテーションを付けるツールとして、SeattleSeq Annotationを紹介しました。ここ
ちょっと調べると、ほかにもいろいろありますね。
今日は定番?のEnsembl です。
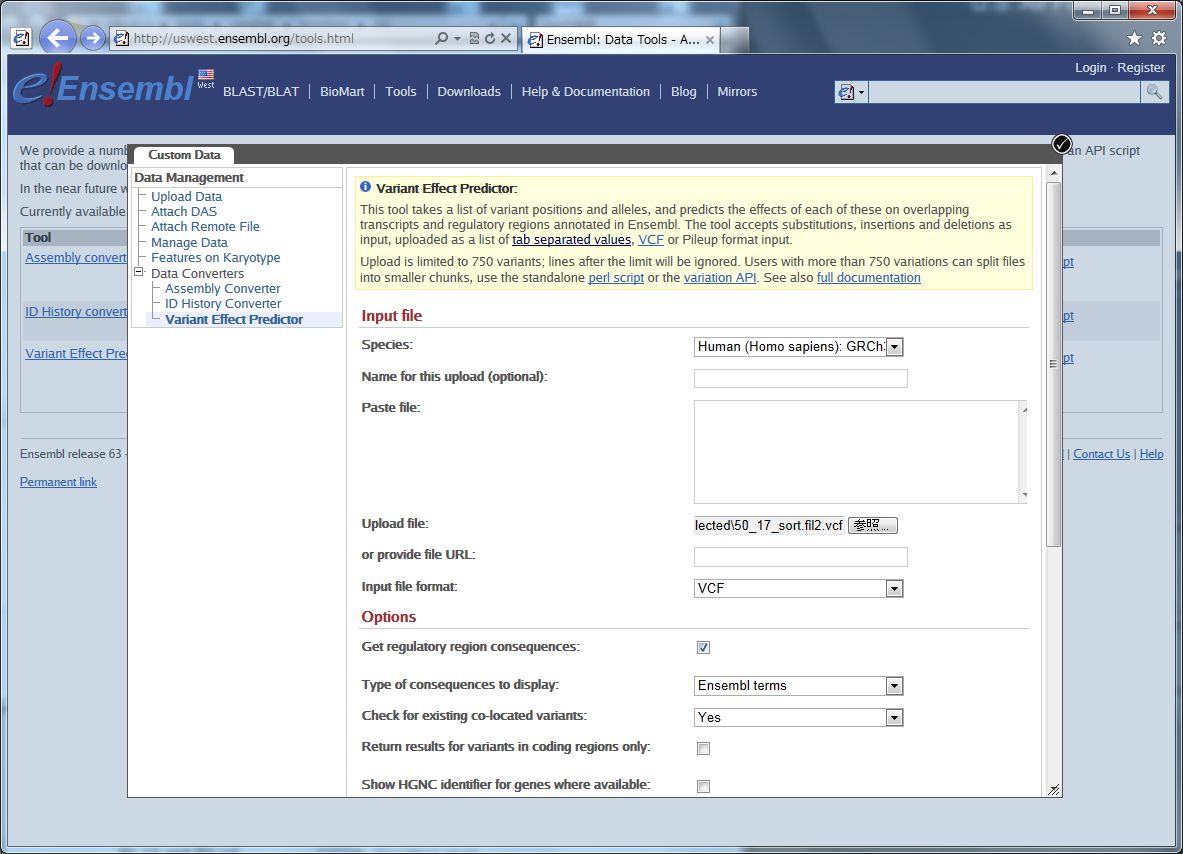
その中の、Variant Effect Predictor というウェブツールです。

Species は、ここではヒト。 ゲノムバージョンと一緒に選びます。
ファイルの場所を指定して、フォーマット(VCF)を選びます。
もう気づいたかもしれませんが、私はVCFを読みこめるツールが好きです。
VCFは変異レポートファイルのデファクトスタンダード。
書き込める自由度も高く、ほぼ満足です。
VCF自体を人間が読むことはあまり無いでしょうが。

追加するアノテーションを選びます。
ここからはヒトのデータのみです。
Non-Synonymous SNP(アミノ酸コーディング領域で起こる一塩基変異で、コードされるアミノ酸を置換するもの)について、その影響を予測するために、SIFT、PolyPhen、の2種類のスコアをアノテーションします。
その下のフィルタリングオプションは、マイナー・アレル・フリークエンシー(MAF)で、データをカットできます。
Nextを押して実行すると、検索が始まります。
こんな画面がでたら終了。
HTMLを「別のタブまたは別画面」で開いてみましょう。 そのまま開くと前に戻れないので注意!

HTMLのレポート画面です。
Ensembl-IDとか、SNVの遺伝子上の場所、変異のアノテーション、アミノ酸変異があるものはその機能への影響度合い、などがリストで表示されます。


さて、HTMLのほかに、テキストでダウンロードもできます。
これは後で編集するのに便利でしょう。
もうひとつは、
Ensemblのゲノムビューワーへのリンク。私は使ったことはことがないですが。 こんな感じでSNVの位置がわかります。

このEnsembl Variant Effect Predictorですが、残念ながらウェブ上で行うにはデータ量MAX780変異と、制限があるそうです。
それ以上の変異を解析するには、Perl APIという手があります。
ちょっと調べると、ほかにもいろいろありますね。
今日は定番?のEnsembl です。
その中の、Variant Effect Predictor というウェブツールです。
Species は、ここではヒト。 ゲノムバージョンと一緒に選びます。
ファイルの場所を指定して、フォーマット(VCF)を選びます。
もう気づいたかもしれませんが、私はVCFを読みこめるツールが好きです。
VCFは変異レポートファイルのデファクトスタンダード。
書き込める自由度も高く、ほぼ満足です。
VCF自体を人間が読むことはあまり無いでしょうが。

追加するアノテーションを選びます。
ここからはヒトのデータのみです。
Non-Synonymous SNP(アミノ酸コーディング領域で起こる一塩基変異で、コードされるアミノ酸を置換するもの)について、その影響を予測するために、SIFT、PolyPhen、の2種類のスコアをアノテーションします。
その下のフィルタリングオプションは、マイナー・アレル・フリークエンシー(MAF)で、データをカットできます。
Nextを押して実行すると、検索が始まります。
こんな画面がでたら終了。
HTMLを「別のタブまたは別画面」で開いてみましょう。 そのまま開くと前に戻れないので注意!

HTMLのレポート画面です。
Ensembl-IDとか、SNVの遺伝子上の場所、変異のアノテーション、アミノ酸変異があるものはその機能への影響度合い、などがリストで表示されます。


すぐ上のスクリーンショットには、Coding領域にあるNon-Synonymous SNVがありますね。
SIFTとPolyPhenの値も右にあります。
SIFT=tolerated(0.07); PolyPhen=benign(0.16)
このうちPolyPhenの値とDescriptionについては以前SeattleSeqのところでちょっと書きましたね。
SIFTについてはまた次回。さて、HTMLのほかに、テキストでダウンロードもできます。
これは後で編集するのに便利でしょう。
もうひとつは、
Ensemblのゲノムビューワーへのリンク。私は使ったことはことがないですが。 こんな感じでSNVの位置がわかります。

このEnsembl Variant Effect Predictorですが、残念ながらウェブ上で行うにはデータ量MAX780変異と、制限があるそうです。
それ以上の変異を解析するには、Perl APIという手があります。
登録:
コメント (Atom)