先月末から何ていうのでしょう、一種のスランプみたいな、そんな気分でしたので。
もう脱しつつあるので、大丈夫です。
昔からGWASなどでSNPを解析しているひとには常識かもしれませんが、NGSが出た最近やっとSNP(SNV)を始めてみようかな、というひとには意外と「目からうろこ」なことがあります。
私もSNPを昔からやっていたわけではないので、たまに論文を読んでいて気づかされることがあります。
で、そういうことを他の研究者と話すと、意外とその人も知らなかったり。
具体的には、NGSを使ってWhole Exomeをやっている解析で、SNVを見つける、そのときのワークフローです。
当たり前のように行っていたフィルタリングのプロセスで、「なぜそのフィルタリングを行うのか」「基本となる考え方・仮定は何か」という基本的なところがおろそかになっていたと。
そこについてメモ書きのようですが、まとめました。
Exome解析の最終目的は、おおよそ、「疾患原因となるSNVの候補を絞り込む」ことになると思います。
そこで、
- ヒトならヒトのExon領域をカバーしたターゲットシーケンスを行い、その領域で十分な厚みを持ってマップされた場所から、SNVを見つけます。 (例:SureSelectなどでターゲットキャプチャーしたあとIlluminaマシンで大量に読み、BWAでヒトゲノムにマッピング、キャプチャー領域にてSamtoolsでSNVを検出)
- 検出されたSNVの場所とリストをもとに、dbSNPなどのデータベースに無いものを抽出
- さらにその中からNon-synonymousのSNV(アミノ酸置換を伴うSNV)のみを抽出
- そして残ったnsSNVのうち、患者複数のサンプルで共通するものを選び出す
それを理解していないでなんとなくパイプライン的に解析を処理していると、後から他の研究者に突っ込まれてタジタジ・・・となってしまう!
以下はSNPをやってたひとには当たり前すぎることを書いていると思います。 どうぞお許しを。
- Exon領域をキャプチャーする大前提は、テーマとしている疾患の変異を、遺伝子のコーディング領域から探そうとしているわけです。 つまりコーディングされない場所にいくら疾患原因SNVがあったとしても、それは見ないことにしよう、としています。
もちろんコーディング領域は重要です。 しかしそれ以外の転写制御領域のSNVも大変重要ですが、これらは一般的なExomeでは解析対象から外されます。 - dbSNPに無い、SNVのみを探すというのは、一見、新規性を探索するのに合理的な方法です。 この疾患原因SNVは非常にレア、珍しいという前提があります。 しかし、データベースに登録されるSNVは、現在爆発的に増え続けていますので、既知であっても自分の研究疾患では新規ということは十分あり得ます。
- Non-synonymousのSNVに絞るということは、アミノ酸置換、あるいはフレームシフト、ミッセンス等を伴うような変異は特に重要だという前提に立っています。 「この疾患の原因は、アミノ酸変異を伴うSNVだ」 という仮定がそこにあります。 そうでない仮定の場合にNon-synonymous SNVに絞るのは適当でないでしょう。
また気をつけるべきは、Alternative Exon(スプライスバリアント)の存在です。 これを無視してSNVを見つけているケースが多いのではないでしょうか。 ゲノムのSNVではなく遺伝子コーディング領域中のSNVとなると、スプライスバリアントが異なればそのSNVの有る無しの意味は大きいはずですよね。 (言葉足らず) - 複数の患者のサンプルで同じSNVを見つけるというのは、そのフェノタイプに共通するSNVを見つけ出すということです。 SNV(原因)があれば必ずそのフェノタイプ(結果)を引き起こすことをComplete Penetranceと言います。 一方、そのフェノタイプ(結果)には必ずそのSNV(原因)が見られるときをComplete Detectance と言います。
つまりこのフィルタリングは、全ての患者に共通するSNVがあるはずだ、という前提に立っているわけです。
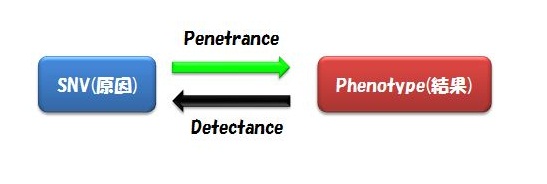
まとめると、上記のワークフローで解析するための前提・仮定は、
「全ての患者に共通するSNVがあって、かつそのSNVを持っていればかなりの確率でその疾患にかかり、そのSNVはとても珍しく、そしてタンパク質のアミノ酸コードを変えることで疾患フェノタイプを引き起こす」
ということになりますでしょうか。
今日は字ばっかりですみませんでした。