accepted_hits.bam, junctions.bed, insertions.bed / deletions.bed というファイルができたとしましょう。

この3ファイルが何かについては前回説明しました。
TopHatは、コマンド実行時に指定したgtfファイルを使って、gtfに書かれている既知の転写配列情報から、Exon Junction を認識してマッピングしてくれるのです。
Exon Junctionを認識するということは、Fusion Geneも特定してくれそうです。
それは、Tophat-fusion というツールがあるのですが、また今度別のときに書きます。
さて、うまくマッピングされ、BAMファイルができました。
では次に、Samtools を使って、BAMファイルを染色体番号順にソートします。
これは後で必要になります。
samtools sort ./tophat_SRP003186_Control/accepted_hits.bam ./tophat_SRP003186_Control/sort_accepted_hits
samtools sort ./tophat_SRP003186_MCF7/accepted_hits.bam ./tophat_SRP003186_MCF7/sort_accepted_hits
samtools sort ./tophat_SRP003186_SKBR3/accepted_hits.bam ./tophat_SRP003186_SKBR3/sort_accepted_hits最後のアウトプットに.bam と書くと、出力が.bam.bam と2重拡張子になるので注意!
samtools index ./tophat_SRP003186_Control/sort_accepted_hits.bam
samtools index ./tophat_SRP003186_MCF7/sort_accepted_hits.bam
samtools index ./tophat_SRP003186_SKBR3/sort_accepted_hits.bam
拡張子.bai というファイルができるはずです。 これがインデックスです。
ついでにソートしたBAMファイルを、SAMフォーマットにしておきましょうか? 別に必要無いですけど、samtools に慣れていないひとは練習がてら。
ちなみにIGVは、SAMファイルも取り込めます。その際には、IGV-toolsでインデックスをつけるひつようがあります。
また、以前紹介した、SAM-Mate というWindows 対応のRNA-SEQ解析ツールは、SAMでないと受け付けてくれません、今のところ。
samtools view ./tophat_SRP003186_Control/sort_accepted_hits.bam -o ./tophat_SRP003186_Control/Control_sort_accepted_hits.samsamtools view ./tophat_SRP003186_MCF7/sort_accepted_hits.bam -o ./tophat_SRP003186_MCF7/MCF7_sort_accepted_hits.sam
samtools view ./tophat_SRP003186_SKBR3/sort_accepted_hits.bam -o ./tophat_SRP003186_SKBR3/SKBR3_sort_accepted_hits.sam
発現変動を見るには、Cufflinksのコマンドのひとつ、Cuffdiff というものを実行します。
ソートしたBAMファイル、既知トランスクリプトのGTFファイルの2つを使います。
cuffdiff -p 4 -v /Path_to_gtf/refGene.gtf ./tophat_SRP003186_Control/sort_accepted_hits.bam ./tophat_SRP003186_MCF7/sort_accepted_hits.bam -o ./cuffdiff/MCF7
cuffdiff -p 4 -v /Path_to_gtf/refGene.gtf ./tophat_SRP003186_Control/sort_accepted_hits.bam ./tophat_SRP003186_SKBR3/sort_accepted_hits.bam -o ./cuffdiff/SKBR3コントロール v.s. MCF-7、コントロール v.s. SK BR-3 の2つの比較ですね。
本当はリプリケートがあると良かったのですが、これはN=1です。
パラメータは単純にデフォルトで行っています。
唯一、CUPを -p 4 にしたことくらいでしょうか。
Cuffdiffのアウトプットファイルは地味で、タブ区切りテキストファイルができるだけです。
もちろん加工すればGenome Viewer上でも表示できますが。
FPKM tracking ファイルと、Differential expression 結果ファイルの2種類があります。
isoforms.fpkm_tracking: Transcript FPKMs
genes.fpkm_tracking: Gene FPKMs. 同じgene_idを持つトランスクリプトのFPKMの積算
(cds.fpkm_tracking: Coding sequence FPKMs.)
(tss_groups.fpkm_tracking: Primary transcript FPKMs)
2) Differential expression 結果ファイル: 各cuffdiffランごとに4ファイル
isoform_exp.diff: Transcript differential FPKM. gene_exp.diff: Gene differential FPKM.
(tss_group_exp.diff: Primary transcript differential FPKM)
(cds_exp.diff: Coding sequence differential FPKM)
このうち、発現変動の結果ファイル、isoform_exp.diff をExcelで開いてみるとこんな感じです。
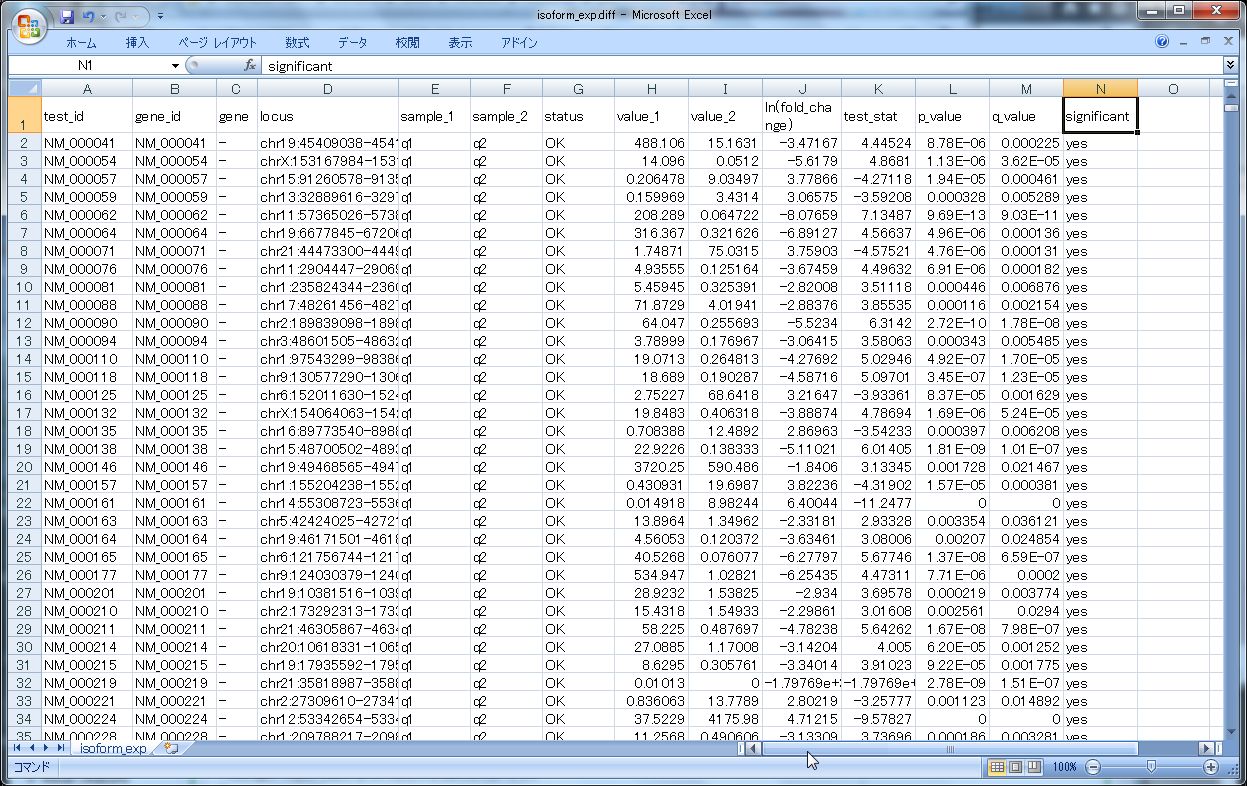
トランスクリプトのIDや名前、ポジション情報が先ずあり、そのあとHカラムくらいから、実際のカウンティングデータがきます。
- FPKMx: FPKM of the gene in sample x
- FPKMy: FPKM of the gene in sample y
- ln(FPKMy/FPKMx):The natural log (自然対数) of the fold change y/x
- test stat: FPKMの変動が有意であるかどうかの統計量
- p value: 補正無しの p-value
- q value: FDR-adjusted p-value
- significant:"yes" or "no" マルチサンプル比較の場合はBenjamini-Hochberg補正後、FDR-adjusted p-valueが、cuffdiffランのパラメータ --FDR (デフォルト:0.05)より小さい場合は、"Yes" 有意だとされる
Ratioは、自然対数のログ表記という点にご注意ください。
また、Excelはsignificant = Yes で絞り込んでいます。
こんな風にして、RNA-Seq の実験データから、発現変動の有意な遺伝子を解析することができるのです。
実際は、パラメータの調整などが必要ですが、感じはわかって頂けたと思います。
このあと考えられる操作は、
- 発現変動の有意だった遺伝子のポジション情報からBedファイルを作り、
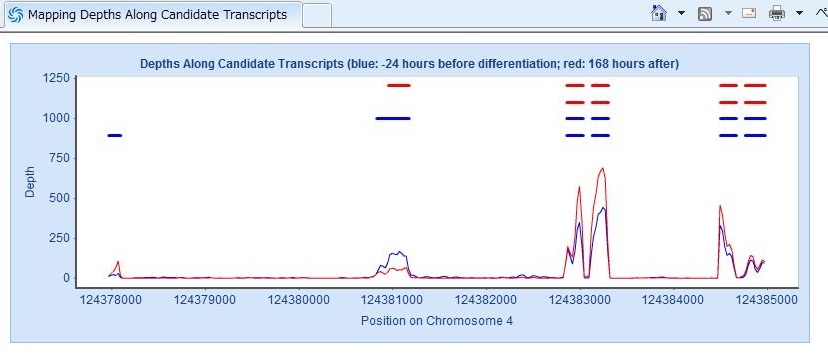
- 先ほどソート済みのBAMファイルをGenome Viewerと一緒にとりこみ、
- ゲノム上で発現の変動と、マッピングの様子を同時に見る
ことでしょうか。 これはまた、Viewerに関する書き込みのときに詳しく説明します。
つづく