accepted_hits.bam, junctions.bed, insertions.bed / deletions.bed というファイルができたとしましょう。

この3ファイルが何かについては前回説明しました。
TopHatは、コマンド実行時に指定したgtfファイルを使って、gtfに書かれている既知の転写配列情報から、Exon Junction を認識してマッピングしてくれるのです。
Exon Junctionを認識するということは、Fusion Geneも特定してくれそうです。
それは、Tophat-fusion というツールがあるのですが、また今度別のときに書きます。
さて、うまくマッピングされ、BAMファイルができました。
では次に、Samtools を使って、BAMファイルを染色体番号順にソートします。
これは後で必要になります。
samtools sort ./tophat_SRP003186_Control/accepted_hits.bam ./tophat_SRP003186_Control/sort_accepted_hits
samtools sort ./tophat_SRP003186_MCF7/accepted_hits.bam ./tophat_SRP003186_MCF7/sort_accepted_hits
samtools sort ./tophat_SRP003186_SKBR3/accepted_hits.bam ./tophat_SRP003186_SKBR3/sort_accepted_hits最後のアウトプットに.bam と書くと、出力が.bam.bam と2重拡張子になるので注意!
samtools index ./tophat_SRP003186_Control/sort_accepted_hits.bam
samtools index ./tophat_SRP003186_MCF7/sort_accepted_hits.bam
samtools index ./tophat_SRP003186_SKBR3/sort_accepted_hits.bam
拡張子.bai というファイルができるはずです。 これがインデックスです。
ついでにソートしたBAMファイルを、SAMフォーマットにしておきましょうか? 別に必要無いですけど、samtools に慣れていないひとは練習がてら。
ちなみにIGVは、SAMファイルも取り込めます。その際には、IGV-toolsでインデックスをつけるひつようがあります。
また、以前紹介した、SAM-Mate というWindows 対応のRNA-SEQ解析ツールは、SAMでないと受け付けてくれません、今のところ。
samtools view ./tophat_SRP003186_Control/sort_accepted_hits.bam -o ./tophat_SRP003186_Control/Control_sort_accepted_hits.samsamtools view ./tophat_SRP003186_MCF7/sort_accepted_hits.bam -o ./tophat_SRP003186_MCF7/MCF7_sort_accepted_hits.sam
samtools view ./tophat_SRP003186_SKBR3/sort_accepted_hits.bam -o ./tophat_SRP003186_SKBR3/SKBR3_sort_accepted_hits.sam
発現変動を見るには、Cufflinksのコマンドのひとつ、Cuffdiff というものを実行します。
ソートしたBAMファイル、既知トランスクリプトのGTFファイルの2つを使います。
cuffdiff -p 4 -v /Path_to_gtf/refGene.gtf ./tophat_SRP003186_Control/sort_accepted_hits.bam ./tophat_SRP003186_MCF7/sort_accepted_hits.bam -o ./cuffdiff/MCF7
cuffdiff -p 4 -v /Path_to_gtf/refGene.gtf ./tophat_SRP003186_Control/sort_accepted_hits.bam ./tophat_SRP003186_SKBR3/sort_accepted_hits.bam -o ./cuffdiff/SKBR3コントロール v.s. MCF-7、コントロール v.s. SK BR-3 の2つの比較ですね。
本当はリプリケートがあると良かったのですが、これはN=1です。
パラメータは単純にデフォルトで行っています。
唯一、CUPを -p 4 にしたことくらいでしょうか。
Cuffdiffのアウトプットファイルは地味で、タブ区切りテキストファイルができるだけです。
もちろん加工すればGenome Viewer上でも表示できますが。
FPKM tracking ファイルと、Differential expression 結果ファイルの2種類があります。
isoforms.fpkm_tracking: Transcript FPKMs
genes.fpkm_tracking: Gene FPKMs. 同じgene_idを持つトランスクリプトのFPKMの積算
(cds.fpkm_tracking: Coding sequence FPKMs.)
(tss_groups.fpkm_tracking: Primary transcript FPKMs)
2) Differential expression 結果ファイル: 各cuffdiffランごとに4ファイル
isoform_exp.diff: Transcript differential FPKM. gene_exp.diff: Gene differential FPKM.
(tss_group_exp.diff: Primary transcript differential FPKM)
(cds_exp.diff: Coding sequence differential FPKM)
このうち、発現変動の結果ファイル、isoform_exp.diff をExcelで開いてみるとこんな感じです。
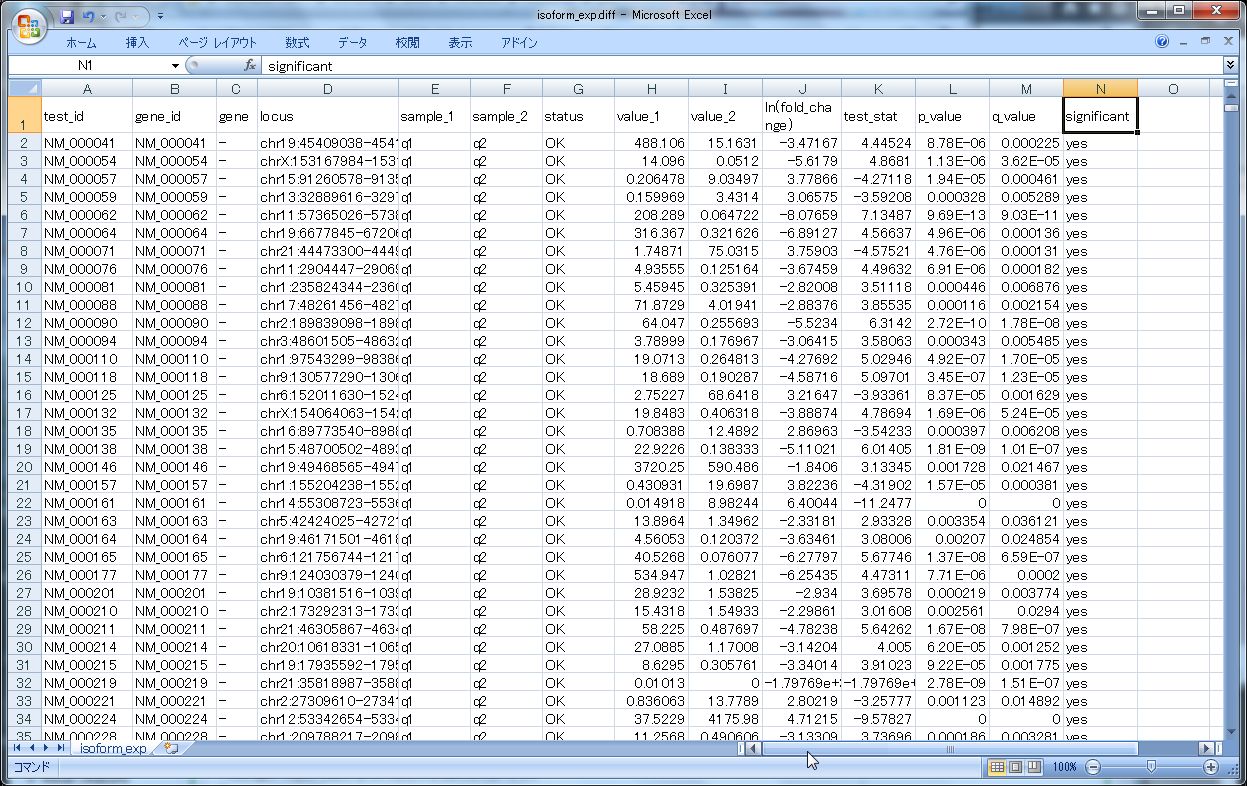
トランスクリプトのIDや名前、ポジション情報が先ずあり、そのあとHカラムくらいから、実際のカウンティングデータがきます。
- FPKMx: FPKM of the gene in sample x
- FPKMy: FPKM of the gene in sample y
- ln(FPKMy/FPKMx):The natural log (自然対数) of the fold change y/x
- test stat: FPKMの変動が有意であるかどうかの統計量
- p value: 補正無しの p-value
- q value: FDR-adjusted p-value
- significant:"yes" or "no" マルチサンプル比較の場合はBenjamini-Hochberg補正後、FDR-adjusted p-valueが、cuffdiffランのパラメータ --FDR (デフォルト:0.05)より小さい場合は、"Yes" 有意だとされる
Ratioは、自然対数のログ表記という点にご注意ください。
また、Excelはsignificant = Yes で絞り込んでいます。
こんな風にして、RNA-Seq の実験データから、発現変動の有意な遺伝子を解析することができるのです。
実際は、パラメータの調整などが必要ですが、感じはわかって頂けたと思います。
このあと考えられる操作は、
- 発現変動の有意だった遺伝子のポジション情報からBedファイルを作り、
- 先ほどソート済みのBAMファイルをGenome Viewerと一緒にとりこみ、
- ゲノム上で発現の変動と、マッピングの様子を同時に見る
ことでしょうか。 これはまた、Viewerに関する書き込みのときに詳しく説明します。
つづく
初めまして。最近GlaxyをもちいたRNA-seqのバイオインフォマティクス解析をはじめまして、いつも貴ブログの内容を参考にさせていただき大変助かっております。ありがとうございます。
返信削除もしお手数でなければ一点お教え頂けないでしょうか。
コントロール群N=3, 実験群N=3、計6サンプルからRNAを抽出し、イルミナ社のnextseqにてRNAseqを行い、取得したデータをもちい、現在解析をおこなっております。cuffdiffによる発現量比較の解析は何とか完了できたのですが、FPKMの値が各郡1つずつしか出ず、個別の各サンプルのFPKMの値がどうしても算出できず(6サンプルの各個別のFPKMの値が出ない)苦戦しております。ヒートマップ作成時に各サンプルのFPKMの値が必要になってくると思いますが、個別でFPKM値を算出する方法はご存知でしょうか。
大変恐縮ではございますが、もしお手数でなければお教えいただけますと幸いです。どうかよろしくお願いいたします。