ダンベル型のSMRTBellというそうです。
Travers et al., A flexible and efficient template format for circular consensus sequencing and SNP detection (2010). Nucleic Acids Research 38, e159.

左端の、輪っかの部分がシングル鎖になっていますね。ここにプライマーがくっつきます。
ポリメラーゼは、DNAの二重鎖のところを押しのけて、プライマーの先に、相補鎖に正しい塩基を合成していきます。
テンプレートは、ずーっといくと一周しますね。
そうしたらまた、Bのように、今合成した通りにもう一度合成をするのです。
つまり、インサートの長さに依存することなく、何度でもシーケンスが可能です。
これがダンベル型テンプレート。
このままの形がスタンダードです。
インサートの長さがものすごーく長い(数10kbとか)とき、全部読むことはできないので輪っかの近いところだけを読むことになります。 これはMait-Pairのような感じです。
Strobeと呼ぶそうです。
もうひとつはインサートの長さは適度で、テンプレートを何周も読む方法。
何度も何度も同じ配列を読むことになるので、精度は上がります。
Circular Consensusと呼ぶそうです。
ここから先は、スタンダードのテンプレートの話です。
SMRTでは、インサートを全部読むので、Read長=インサート長、になります。(SMRTについては前回のブログを参照)
しかし、全部読み切れなかったインサートもあるでしょう。
そんな、「途中まで読めたインサート」配列が混じっている状態のReadのことを、Subreadと呼ぶそうです。
Subreadがいくつか集まってRead(=インサート配列)を形成する、ということです。
ですから、SMRTで読んだ解析結果には必ず、
Subread の平均の長さ
Read の平均の長さ、95パーセンタイルの長さ、・・・
など、Subread とReadの両方の情報があるのが普通でしょう。
前回紹介した、Expression Analysis社のWebセミナーで紹介していたのは、SMRTで大腸菌のゲノムを読んだときのデータ解析についてです。
深夜2時過ぎのことでしたので私の記憶が正しく無い箇所もあるかもしれません。
2kbと6kbの2種類のインサートを読んでいました。
SMRTのひとつのセル(フローセルのような単位)で、34~108 MbのMappableなデータが出力されました。
平均1500塩基長、プロトコルを改良すると2700塩基長を一度に読めていました。
特徴のひとつは、大腸菌ゲノムに対するカバレージが一定だったことです。
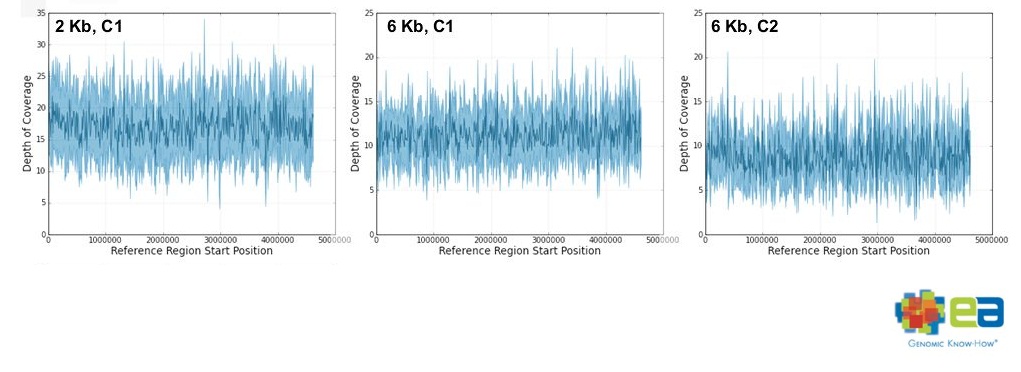
それが比較的無くカバレージが一定で、
2kbインサートが6.4~17.1カバレージ
6kbインサートが6.7~11.2カバレージをゲノム全体で保持していたそうです。
精度ですが、Subreadのレベルの精度は85%程だったそうです。
これがSMRTの精度だと一般的に言われると、15%は間違い? 結構高いね、と思われそうですが、SubreadではなくRead、あるいは何度も繰り返して読んだ時のConsensusともなると、96.46~99.39%まで向上していたそうです。
何度も読むというのは、賢い方法です。
ダンベル型テンプレートならでは?
ランニングコストがどれくらいなのかはわかりませんが、もっと汎用的に使われる時はもうすぐ来るのでしょうか。
個人的には、機械が大きすぎ! って思います。

もうちょい、コンパクトにならないのか・・・





